
Abstract
The present article summarizes progress in research on silicon clusters with encapsulated metal atoms, and specifically focuses on the recent identification of magnetic silicon fullerenes. Considering that C\(_{20}\) forms the smallest known fullerene, the Si\(_{20}\) cluster is of particular interest in this context. While the pure hollow Si\(_{20}\) cage is unstable due to the lack of \(sp^2\) hybridization, endohedral doping with a range of metal atoms has been considered to be an effective way to stabilize the cage structure. In order to seek out suitable embedded atoms for stabilizing Si\(_{20}\), a broad search has been made across elements with relatively large atomic radius. The rare earth elements have been found to be able to stabilize the Si\(_{20}\) cage in the neutral state by forming R@Si\(_{20}\) fullerene cages. Among these atoms, Eu@Si\(_{20}\) has been reported to yield a stable magnetic silicon fullerene. The central europium atom has a large magnetic moment of nearly 7.0 Bohr magnetons. In addition, based on a stable Eu\(_2\)Si\(_{30}\) tube, a magnetic silicon nanotube has been constructed and discussed. These magnetic silicon fullerenes and nanotubes may have potential applications in the fields of spintronics and high-density magnetic storage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
How to Stabilize Silicon Fullerene
Fullerenes, first reported by Curl, Kroto, and Smalley [1], are highly symmetric cage-shaped molecules, and have been thoroughly studied during the last three decades. In the fullerenes, all the atoms are C atoms and they form a hollow sphere comprised of pentagonal and hexagonal rings. Since the first preparative-scale isolation of the C\(_{60}\) molecule in 1990 by Krätschmer and Huffman [2], who found that it was possible to isolate fullerenes in bulk quantities from carbon soot either by dissolving them in benzene or by subliming them at 400 °C, a wide variety of chemically modified fullerenes has been synthesized, and outstanding structural, magnetic, superconducting, electrochemical, and photophysical properties have been reported [3–5]. The carbon based nanomaterials not only display novel mechanical, electronic, optical, and super conducting properties themselves, but they have also served as vehicles for the synthesis of new nano-structures with the insertion of atoms and clusters. The new field of fullerene chemistry is leading to many novel derivatives, some of which may have additional, extraordinary, optical and electronic properties [6].
Silicon and carbon belong to the same group, group IV, of the periodic table, and they have similar valence electron configurations. Silicon is also the most widely used semiconducting material in the microelectronic industry. With the tremendous development of smaller and smaller silicon semiconductor devices, there is currently great interest in studying silicon clusters, and they have attracted extensive experimental and theoretical attention [7, 8]. All these points raise the question as to whether carbon can be substituted by silicon in its nanostructures. The first problem is therefore whether or not the fullerene Si\(_{60}\) cluster is stable.
Menon and Subbaswamy[9] have used Harrison’s parameters for generalized tight-binding molecular dynamics to study the structure of the fullerene Si\(_{60}\) cluster. They found that icosahedral Si\(_{60}\) has metallic character with a partially filled fivefold level as the highest occupied molecular orbit (HOMO). Consequently, Jahn-Teller distortion can change the icosahedral structure to a lower symmetry structure with no changes to the threefold coordination. Piqueras et al. [10] and Nagase and Kobayashi [11] have also reported on the electronic structure of icosahedral Si\(_{60}\). Based on symmetry-restricted optimization, they found that icosahedral Si\(_{60}\) to occupy an energy minimum, in which the fivefold HOMO is completely filled. This is different from the results obtained using parameter dependent tight-binding molecular dynamics, and they found a value for the gap between the HOMO and the lowest unoccupied molecular orbit (LUMO) that was clearly too large, up to 6.69 eV for the fullerene C\(_{60}\) and 4.62 eV for the icosahedral Si\(_{60}\). Later, using density functional theory with the local density approximation, Gong and Zheng [12] studied the electronic structure and stability of a fullerene-like Si\(_{60}\) cluster. They found that the icosahedral structure of Si\(_{60}\) has a small HOMO-LUMO gap and two distinct bond lengths, and that the electronic structure near the Fermi level is similar to that of a C\(_{60}\) cluster. Since the icosahedral Si\(_{60}\) cluster has a quite large cage, and has a small binding energy, it may be a meta-stable structure and may collapse into a more compact structure as the smaller cluster does. In 2007, an investigation by Zhao et al. [13] using a genetic algorithm and density-functional theory showed that an endohedral fullerene of Si\(_{12}\)@Si\(_{48}\) was the more stable than and energetically preferred to the hollow \(I_h\) cage by 12.371 eV. Gong et al. have tried to stabilize icosahedral Si\(_{60}\) by doping with a stable C\(_{60}\) cluster, and found that the C\(_{60}\)@Si\(_{60}\) cluster can be stable with a completely filled HOMO and a sizable gap at the Fermi level. The prediction that C\(_{60}\)@Si\(_{60}\) could be energetically stable has in turn led to many experimental investigations [14–17]. Unfortunately, it has been found experimentally that Si\(_{60}\) is unlikely to wet the surface of C\(_{60}\) [15]. Using first-principles calculations, Sun et al. [18] carried out a comprehensive search for stable geometries of bare Si\(_{60}\) and Si\(_{60}\) supported on a C\(_{60}\) fullerene. They showed that Si\(_{60}\) and C\(_{60}\)@Si\(_{60}\) clusters are both unstable if a nested fullerene-like cage structure is assumed. However, they found that the Si\(_{60}\) cage could be stabilized by including within it, as endohedral units, small “magic clusters” such as Al\(_{12}\)X (X = Si, Ge, Sn, Pb) and Ba@Si\(_{20}\).
In summary, these many clues have motivated a search for a strategy to stabilize silicon cages. As silicon has a larger atomic radius and lower electronegativity than carbon, and lacks \(sp^2\) bonding, pure hollow silicon cages are unstable, and much attention has been focused on how to stabilize silicon fullerenes. An effective approach is to encapsulate guest atoms such as transition metals (TMs). It is now thought possible that silicon cages with encapsulated metal atoms may lead to applications that could match or even exceed those expected for carbon fullerenes. Thus, encapsulated Si cages could lead to an entirely new range of applications of Si-based nanomaterials.
Cage-Like Clusters of Silicon with Encapsulated Metals
In recent years, several significant experimental studies have reported the formation of metal-doped silicon clusters [19–22]. Using a laser vaporization supersonic expansion technique, the production and study of small mixed TM@Sin clusters (TM = Cr, Mo, W; \(n < 19\)) were first reported by Beck [19, 20]. In this excellent experimental work, Beck observed the formation of silicon clusters containing TM atoms, and by subjecting them to photo-fragmentation, found that the doped clusters were more stable than the bare silicon clusters of the same size. They found Si\(_{15}M\) and Si\(_{16}M\) to be the dominant species formed, and also found them to be stable against dissociation. Hiura et al. [21] used an ion trap to study the reaction of TM atoms with silane (SiH\(_4\)) producing a series of dehydrogenated MSi\(^+_n\) cluster ions with n = 14, 13, 12, 11 and 9. This work showed that the metal atom is endohedral and stabilizes the Si polyhedral cage. Koyasu et al. [22] studied the electronic and geometrical structures of mixed-metal silicon TM@Si\(_{16}\) (TM = Sc, Ti, and V) using mass spectrometry and anion photoelectron spectroscopy. They found that neutral TiSi\(_{16}\) clusters had a closed-shell electron configuration with a large gap between the HOMO and LUMO orbitals. Recently, based on photoelectron spectroscopy and theoretical analysis using density functional theory, Huang et al. have discovered a Si-based ferrimagnetic cluster (V\(_3\)Si\(_{12}\)) with a symmetric cage configuration [23]. Further, they found that this ferrimagnetic cage can be extended into a ferrimagnetic nanotube.
Motivated by these experimental advances, there has been a variety of first-principles investigations of TM-doped silicon clusters [24–36], in which, the TM clusters have usually been found to have non-zero magnetic moments [37]. Kawazoe and colleagues [24–28] reported a series of systematic investigations of TM-doped silicon clusters using ab initio pseudopotential plane-wave calculations within the generalized gradient approximation (GGA) for the exchange-correlation energy. They found that silicon forms fullerene-like MSi\(_{16}\) (M = Hf, Zr) or cubic TMSi\(_{14}\) (TM = Fe, Ru, Os) cage clusters, depending on the size of the metal atom. Lu and Nagase [29] investigated TM@Si\(_n\) (TM = W, Zr, Os, Pt, Ru, Fe, Co; n = 8–20) using spin-unrestricted hybrid density-functional theory with the B3LYP exchange-correlation functional, and found that the formation of the endohedra structure strongly depended on the size of the silicon cluster. In addition, based on the symmetry of the structures and the d-band filling, Mpourmpakis et al. [30, 31] gave an analysis of the structure of the silicon clusters using ab initio pseudopotential plane-wave calculations and DFT within the GGA. Ma et al. [32, 33] investigated small size TM@Si\(_n\) (TM = Fe, Co; n = 1–13) clusters and found, using DFT combined with a genetic algorithm, that the stability of the metal-doped clusters was enhanced with increasing cluster size. Small size NiSi\(_n\) (n = 1–8) clusters were investigated computationally under the constraint of well-defined symmetries at the B3LYP level by employing a pseudopotential method in conjunction with the Los Alamos double zeta basis sets [34]. Koukaras et al. [35] have also studied the Ni@Si\(_{12}\) cluster in detail and identified a new lower ground-state structure using all-electron ab initio calculations in the framework of DFT with the B3LYP function which includes hybrid nonlocal exchange and correlation. The configurations and electronic structure of Si\(_n\)Ni (n = 1–14) clusters have been calculated in the framework of all-electron density-functional theory [36]. The calculated results showed that the Ni atom prefers to occupy a surface site when \(n <\) 9, but for clusters with n = 9–14, the Ni atom starts to encapsulate in the cage. In addition, it was found that there was a peak in the size dependence of embedding energy for Si\(_n\)Ni clusters at n = 12, implying that the Si\(_{12}\)Ni cluster is the most stable structure. The result is that the doped Ni atom both enhances the stability of silicon clusters and helps to form the Ni-encapsulating Si cage. In addition, the geometries, stabilities, and electronic properties of YSi\(_n\) (n = 1–6) [38], TiSi\(_n\) (n = 2–15) [39], ZrSi\(_n\) (n = 1–16) [40], TaSi\(_n\) (n = 1–13) [41], CrSi\(_n\) (n = 1–6) [42], MoSi\(_n\) (n = 1–6) [43], WSi\(_n\) (n = 1–6, 12) [44], ReSi\(_n\) (n = 1–12) [45] and IrSi\(_n\) (n = 1–6) [46], have been studied within the context of first-principles calculations. Within the framework of all-electron density functional theory, the configurations, stabilities, and electronic structures of Si\(_{15}\) and Si\(_{16}\) cages with encapsulated 3d transition metal atoms, M@Si\(_{15}\) and M@Si\(_{16}\) (M = Sc, Ti, V, Cr, Mn, Fe, Co, or Ni), have been investigated systematically [47]. The results show that Ti@Si\(_{16}\) and Ti@Si\(_{15}\) have the largest embedding energies in this series and also have relatively large HOMO-LUMO gaps. This suggests that the titanium atom is an ideal guest for Si\(_n\) (n = 15, 16) cages as far as stability is concerned. The Mn atom is found to have a large spin moment even when encapsulated, while the spin moments of Ti, Cr, Fe, and Ni are entirely quenched upon doping into Si\(_{15}\) and Si\(_{16}\) cage clusters. All these theoretical results indicate that the encapsulation of TM atoms in the larger-sized silicon clusters contributes to enhancing the stability of pure silicon clusters and simultaneously brings forth a variety of novel behaviors. This is largely because the TM atom can saturate dangling bonds on the silicon cage surface.
In addition to the general interest in TM-doped silicon clusters, a number of studies have been devoted to group IB metals (Cu, Ag, and Au). Gold, as a good conductor of heat and electricity and one of the most important elements in the periodic table, is the noblest of all metals and has been prized throughout history for its beauty and resistance to corrosion [48]. On the experimental side, the AuSi silicide was first reported by Barrow et al. [49] early in 1964. Recently, the atomic and electronic structures of Au\(_5M\) (M = Na, Mg, Al, Si, P, S, and Au) clusters [50] have been investigated using DFT-GGA calculations. They observed a structural transition from planar to nonplanar configurations depending on the nature of the interaction with different impurity elements. The Al, Si, P impurities prefer nonplanar geometries for Au\(_5\)M clusters, while Na and Mg yield planar geometries. Sun et al. [51] examined the stability of the Si@Au\(_{16}\) cluster using an ab initio simulated annealing method and they found that the Si atom binds to the exterior surface of the Au\(_{16}\) cage in the lowest-energy structure. Results for small clusters were calculated by Kiran et al. [52] who reported calculations on a series of Si–Au clusters, SiAu\(_n\) (n = 2–4). The results showed that the Au atoms behave like H in their bonding to Si. Later, a combined photoelectron spectroscopic and theoretical study was carried out on the geometrical and electronic structures of Si\(_2\)Au\(^-_2\) and Si\(_2\)Au\(^-_4\), and their corresponding neutral species [53]. It was found that the most stable structures for both Si\(_2\)Au\(_2\) and Si\(_2\)Au\(^-_2\) had \(C_{2V}\) symmetry, in which each Au bridges the two Si atoms. For Si\(_2\)Au\(^-_4\), two nearly degenerate structures with two bridges, one structure in a cis (\(C_{2h}\)) and the other in a trans (\(C_{2V}\)) configuration were found to be the most stable isomers. In the neutral potential energy surface for Si\(_2\)Au\(_4\), an isomer with a single bridge was found to have the global minimum in energy. Xiao et al. [54] have studied the geometric, energetic, and bonding properties of CuSi\(_n\) (n = 4, 6, 8, 10, and 12) clusters in both neutral and charged states using a hybrid density functional method (B3LYP). They found that the Si\(_n\) frameworks of most isomers of CuSi\(_n\) adopt the geometries of the ground-state or low-lying isomers of Si\(_n\) or Si\(_{n+1}\) , with Cu at various substitutional or adsorption sites. Several cage-like structures with Cu at the central site were found for CuSi\(_{10}\) and CuSi\(_{12}\). A hexagonal double-chair structure with Cu at the center was identified as the best candidate for the ground state of CuSi\(_{12}\). The structures of AgSi\(_n\) (n = 1–5 [55] and n = 1–13 [56]) were also investigated using first-principles calculations. They found that AgSi\(_n\) clusters with n = 7 and 10 isomers are relatively stable isomers and that these clusters prefer to be exohedral rather than endohedral, with the doping leaving the inner core structure of the clusters largely intact. Alternative pathways exist for \(n >\) 7 (except n = 11) in which the Ag–Si cluster dissociates into a stable Si\(_7\) and a smaller AgSi\(_{n-7}\) fragment. The AgSi\(_{11}\) cluster dissociates into a stable Si\(_{10}\) and a small AgSi fragment. It was also found that the doping of an Ag atom significantly decreases the HOMO-LUMO gaps for n < 7. The configurations, stabilities, and electronic structures of AuSi\(_n\) (n = 1–16) clusters have been investigated within the framework of the density functional theory at the B3PW91/LanL2DZ and PW91/DNP levels [57]. The results show that the Au atom begins to occupy the interior site for cages as small as Si\(_{11}\) and for Si\(_{12}\) the Au atom completely falls into the interior site forming an Au@Si\(_{12}\) cage. A relatively large embedding energy and small HOMO-LUMO gap are also found for this Au@Si\(_{12}\) structure indicating enhanced chemical activity and good electronic transfer properties. All these calculations and experimental studies indicate that silicon clusters can be dramatically stabilized by introducing guest atoms, and the choice of the central guest atom becomes a key point in the design of cage clusters and in their resultant chemical behavior.
Eu@Si\(_{20}\): Magnetic Silicon Fullerene
It is known that the smallest carbon fullerene cage is C\(_{20}\) with a dodecahedral structure [58, 59]. In C\(_{20}\), all faces are pentagonal and the bonding becomes more \(sp^3\) like, which makes the synthesis of C\(_{20}\) difficult. However, this bonding pattern is well-suited for Si\(_{20}\). On the other hand, the energetically unfavorable dangling bonds on this cage lead to reconfiguration so that the lowest energy isomer of pure Si\(_{20}\) has a prolate structure based on stacking Si\(_{10}\) tetracapped trigonal prism units, which suggests that the fullerene cage is not the most stable form for Si\(_{20}\) [60–63]. However, in contrast to the intrinsically unstable hollow Si\(_{20}\) fullerene, as pointed out above, endohedral doping with a range of metal atoms has indeed been theoretically predicted to stabilize the cage structure.
Using first-principles calculations, Sun et al. [64] reported that doping with atoms such as Ba, Sr, Ca, Zr etc leads to distorted M@Si\(_{20}\) cages. The relatively small endohedral doping energies that were found are unlikely to stabilize Si\(_{20}\) fullerene. On the other hand, ab initio electronic structure calculations have suggested that thorium should form a nonmagnetic neutral Th@Si\(_{20}\) fullerene with icosahedral symmetry, and that Th may be the only element that can stabilize the dodecahedral fullerene of Si\(_{20}\) [65]. Using ab initio calculations, Kumar et al. [66] showed that the encapsulation of Y, La, and Ac metal atoms stabilized the dodecahedral fullerene anion M@Si\(^-_{20}\) with icosahedral symmetry. Doping of other rare earths was further shown to stabilize magnetic dodecahedral fullerenes Pa@Si\(_{20}\), Sm@Si\(_{20}\), Pu@Si\(_{20}\), and Tm@Si\(_{20}\) with 1 \(\mu _B\), 4 \(\mu _B\), 4 \(\mu _B\), and 3 \(\mu _B\) spin magnetic moments, respectively. This result is in contrast to most previous studies on M-encapsulated Si clusters in which the magnetic moment was found to be completely quenched. A Gd doped fullerene anion was predicted to have a high magnetic moment of 7 \(\mu _B\), and studies of the photoelectron spectra of EuSi\(_n\) cluster anions (n = 3–17) have shown that EuSi\(_{12}\) is the smallest fully endohedral europium-silicon cluster [67]. Using the density functional approach, Wang et al. [68] calculated stabilities and electronic properties of novel transition bimetallic atoms (TMA) encapsulated in a naphthalene-like Si\(_{20}\) prismatic cage, TMA\(_2\)@Si\(_{20}\). They found that the symmetry and electronic state of naphthalene-like TMA\(_2\)@Si\(_{20}\) was significantly affected by the type of encapsulated TMA chosen from the 3d, 4d and 5d series of the periodic table.
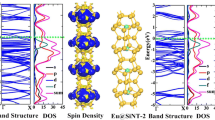
To seek out a suitable embedded atom, a broader search was made through the elements with relatively large atomic radius in the periodic table, including the late alkali metals, the late haloid elements, the 3d and 4d transition metals and the lanthanide series [69]. A rare earth element (R) can stabilize the Si\(_{20}\) cage in the neutral state by forming an R@Si\(_{20}\) fullerene cage. The europium atom has a half-filled f-orbit with a ground state valence-electron configuration of 4\(f^7\)6\(s^2\). The results show that the Eu atom is an ideal “guest” to produce a carbon-like Si\(_{20}\) fullerene due to its large valence shell orbital radii, large atomic magnetic moment and half-filled 4f shell orbital. Figure 1 gives the structure of the stable \(D_{2h}\)-Eu@Si\(_{20}\) fullerene cage, showing that it is a Si\(_{20}\) dodecahedron with complete encapsulation of the europium atom. A vibrational frequency analysis at the PW91 level further confirmed the structural stability of \(D_{2h}\)-Eu@Si\(_{20}\) with no imaginary frequencies. In addition, the validation of the kinetic stability through ab initio molecular dynamic (MD) simulations indicated that the bond structure of Eu@Si\(_{20}\) keeps the same topological structure as \(D_{2h}\)-Eu@Si\(_{20}\). Remarkably, based on the analysis of the electronic structure of the stable Eu@Si\(_{20}\) fullerene cage, a large spin magnetic moment of nearly 7.0 \(\mu _B\) was found for the central europium atom.

The stable Eu@Si\(_{20}\) fullerene. Large ball Eu atom; small ball Si atom.
The stability and electronic structure of the dimers constructed by concatenating two Eu@Si\(_{20}\) subunits were also investigated. Two kinds of dimers were constructed by concatenating two Eu@Si\(_{20}\) subunits with and without a rotation, as shown in Fig. 2. After optimization, it was found that the [Eu@Si\(_{20}\)]\(_2\) dimer kept the Eu@Si\(_{20}\) subunit intact, while distortions occurred for the [Eu@Si\(_{20}\)]\(^`_2\) dimer, which had a rotation between the subunits. The stability was confirmed by comparison with Eu\(_2\)@Si\(_{40}\) clusters obtained by substituting two Eu atoms for two of the eight internal Si atoms of the lowest-energy structure of Si\(_{42}\) [70]. The calculated results show that the constructed [Eu@Si\(_{20}\)]\(_2\) dimer was the lowest-energy structure from the point of view of the binding energy. In the lowest-energy [Eu@Si\(_{20}\)]\(_2\), the central Eu atoms retain a large spin magnetic moment of nearly 7.0 \(\mu _B\) and the spin moments of the two central Eu atoms have the same direction. Subsequently, quasi-one-dimensional nanowires were constructed by concatenating a series of stable Eu@Si\(_{20}\) subunits. Optimizations indicated the existence of a stable pearl necklace nanowire, [Eu@Si\(_{20}\)]\(_{n\rightarrow \infty }\). Figure 2 shows the unit cells of the infinite Eu@Si\(_{20}\) chains. It can be seen that the [Eu@Si\(_{20}\)]\(_{n\rightarrow \infty }\) linear structure obtained by concatenating two Eu@Si\(_{20}\) subunits without a rotation keeps the stable Eu@Si\(_{20}\) subunit intact. Close examination of the band structure and the partial density of states of the [Eu@Si\(_{20}\)]\(_{n\rightarrow \infty }\) nanowire, indicates that it appears to have some of the properties of a direct-gap semiconductor with 0.42 eV energy gap at the \(\Gamma\) point. It is noteworthy that for the pearl necklace nanowire the spin moments of the Eu atoms remain as large as that in the ground-state \(D_{2h}\)-Eu@Si\(_{20}\) fullerene, and the same as for the [Eu@Si\(_{20}\)]\(_2\) dimer. In addition, the spin magnetic moment of the central Eu atom, for each Eu@Si\(_{20}\) subunit of [Eu@Si\(_{20}\)]\(_{n\rightarrow \infty }\), has the same direction. Given the properties of these structures, there may be significant potential for exploiting novel materials based on Eu@Si\(_{20}\) in spintronics devices or in high-density magnetic storage.

The initial and optimized structures of [Eu@Si\(_{20}\)]\(_2\) and [Eu@Si\(_{20}\)]\(^`_2\) dimers and the unit cells of the two optimized pearl necklace nanowires, [Eu@Si\(_{20}\)]\(_{n\rightarrow \infty }\) and [Eu@Si\(_{20}\)]\(^`_{n\rightarrow \infty }\)
Eu\(_2\)Si\(_{30}\): Magnetic Silicon Nanotube
As outlined above, research on the stabilization of metal-encapsulated silicon fullerenes is still an open topic and calls for further discussion. On the basis of a combined photoelectron spectroscopy and first-principles density functional study of Si\(_n\) (n = 20–45), Bai et al. [71] reported that pure silicon clusters exist with a prolate-to-near-spherical shape transition at n = 27, and a half-metallic silicon nanotube (SiNT) with a hybrid atomic chain of Mn and Co was found [72]. The origin of the stability of this silicon nanotube was further explored. A europium-encapsulating magnetic silicon nanotube, Eu\(_2\)@Si\(_{30}\), has also been identified using density functional theory [75].
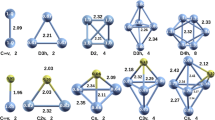
We first discuss the stability of Eu\(_2\)@Si\(_{30}\). To seek out a stable structure for Eu\(_2\)@Si\(_{30}\), two series of initial cage-like Eu\(_2\)@Si\(_{30}\) structures were constructed by substituting Eu at two of the inner sites of stable Si\(_{32}\) [73, 74]. In addition to these cage-like structures, a tubular structure was handcrafted as well. After optimization, 14 low-lying structures of the Eu\(_2\)Si\(_{30}\) cluster were obtained, as shown in Fig. 3. Compared with the initial structures, the silicon cages were propped up by the two Eu atoms. The most stable structure was an irregular prolate silicon fullerene cage (I-a in Fig. 3). This configuration had \(C_1\) symmetry, and the HOMO-LUMO gap was 0.53 eV at the GGA-PW91 level. The structural stability of structure I-a was also confirmed by vibrational frequency analysis. For the I-a isomer, there were no imaginary frequencies and the frequency of the largest intensity peak corresponds to 402 cm\(^{-1}\). The second low-lying isomer, i.e., the “Tube” in Fig. 3, is very attractive due to its large total spin moment of 10 \(\mu _B\) and relatively high stability with only a 0.27 eV higher total energy than the ground state I-a. The Tube structure consists of twelve silicon pentatomic rings and five hexatomic rings and has a \(D_{5h}\) symmetry tubular structure with the two Eu atoms completely encapsulated. Vibrational frequency analysis and ab initio molecular dynamics simulations showed that the Tube isomer is kinetically and thermodynamically stable. Vibrational frequency analysis also showed no imaginary frequencies for this Tube isomer and gave the two higher intensity frequencies as 167.4 and 507.5 cm\(^{-1}\).

Optimized structures of Eu\(_2\)Si\(_{30}\). The binding energy values (eV) beneath each isomer are the relative energies with respect to the lowest lying isomer (isomer I-a)
Further analysis of the electronic structure of the Eu\(_2\)Si\(_{30}\) cluster demonstrates the role that the rare-earth element plays in stabilizing the silicon fullerenes. Figure 4 shows the deformation electronic density of the most stable Si\(_{30}\) isomer [73], the tubular Si\(_{30}\) (removing the two Eu atoms of the Tube structure) and the two Eu\(_2\)@Si\(_{30}\) isomers (I-a and Tube). With encapsulation of the two Eu atoms, there is an obvious decrease in the sp 3-like hybridization and an increase of the sp 2-like hybridization. Upon doping of the two central Eu atoms forming the tube structure (Tube), the same trend can be seen between the tubular Si\(_{30}\) and the Tube structure with encapsulated Eu. From the deformation electron density of the tubular Si\(_{30}\) in Fig. 4b, it can be seen that the Si atoms in the second and fifth layers show a slight sp 3-like hybridization, while the other silicon atoms have sp 2-like hybridizations. For the Tube structure of Eu\(_2\)Si\(_{30}\), there is a reduction of the sp 3-like hybridization for the 10 Si atoms in the second and the fifth layers, but a significant increase in the sp 2-like hybridization. All 30 Si atoms of the Tube structure are the same, in that each has three neighbors, and the three lobes from each Si atom form \(\sigma\) bonds with the three neighboring Si atoms, indicating the characteristics of sp 2-like hybridization. All these results demonstrate that the europium atom is crucial for stabilizing the tubular Si\(_{30}\) through the sp 2-like hybridization induced by the central Eu atoms.

The deformation electronic density and configuration of a Si\(_{30}\), b tubular Si\(_{30}\), c I-a, and d tube. The isosurface is set to 0.022 e/\({\AA }^3\)
The most surprising finding is the high spin magnetic moment of the Tube structure. On the basis of Mulliken population analysis for the Tube structure, it can be seen that both of the central Eu atoms have a spin-up magnetic moment of 6.95 \(\mu _B\), resulting in a total spin moment of 10.00 \(\mu _B\). However, the total magnetic moment of I-a, the most stable Eu\(_2\)Si\(_{30}\) isomer, is zero, even though there is a slightly larger absolute magnetic moment of 7 \(\mu _B\) for each Eu atom, one spin-up and the other spin-down. Similar to the predictions and speculation of Daedalus, this magnetic Eu\(_2\)Si\(_{30}\) Tube, plus its relatively high stability, can be considered as an embryo for the longer magnetic SiNTs. A periodic polymer composed of the magnetic Eu\(_2\)Si\(_{30}\) Tube subunits was built by hand and a stable SiNT with a quasi-chain of Eu atoms in the center was obtained. The unit cell of the new SiNT retains the topological structure of the Eu\(_2\)Si\(_{30}\) Tube, and each of the central Eu atoms retains a large spin-up magnetic moment of 6.99 \(\mu _B\), with the result that the value of the total spin moment for the unit cell is 12.00 \(\mu _B\). When the unit cell is expanded to twice the size, the total spin magnetic moment is 26 \(\mu _B\), i.e., 13 \(\mu _B\) per unit cell, making this spin configuration energetically favored over the antiparallel spin configuration of the Eu atoms. Thus, this SiNT, as expected, is a magnetic SiNT, in which Eu atoms retain a large spin moment. Detailed analysis of the electronic properties shows that, similar to the case of Eu\(_2\)Si\(_{30}\) Tube, the SiNT has an obvious hybridization of Si s-p and Eu f orbitals. Analysis of the band structure shows that this magnetic SiNT is metallic, with several bands in the vicinity of the Fermi level that ensure a significant carrier density. The bands are degenerate at the \(\Gamma\)-point, and they are dispersive along the \(\Gamma\)-X axis, which implies that the effective mass of the charge carriers should be small. Hence, this magnetic SiNT could have potential as a metallic connection in electronic devices.
Conclusions
To summarize, the magnetic silicon fullerenes including silicon fullerenes, metal-encapsulating silicon clusters, and the recently identified magnetic silicon fullerenes and nanotubes have been reviewed. Pure hollow silicon fullerene is unstable due to the lack of \(sp^2\) hybridization. Endohedral doping with rare earth elements, especially europium atoms, has been found to stabilize the Si fullerene by forming an Eu@Si\(_{20}\) cage-like structure and a Eu\(_2\)Si\(_{30}\) tube-like structure. More importantly, both display magnetic properties. Based on the magnetic Eu\(_2\)Si\(_{30}\) tube, a metallic magnetic silicon nanotube was obtained in further calculations, which may have potential applications in spintronics and high-density magnetic storage. Despite the advances discussed above, the study of magnetic silicon fullerenes and nanotubes remains a field with many unresolved issues and many new opportunities. So far, most experiments have been carried out on elemental clusters. It would be desirable to confront the theoretical predictions regarding magnetic silicon fullerenes with experimental data. From the applications point of view, it is important and challenging to explore larger sized magnetic silicon fullerenes and nanotubes.
References
H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl, and R. E. Smalley (1985). Nature 318, 162.
W. Kratschmer, L. D. Lamb, K. Fostiropoulos, and D. R. Huffman (1990). Nature 347, 354.
A. Lappas, K. Prassides, K. Vavekis, D. Arcon, R. Blinc, P. Cevc, A. Amato, R. Feyerherm, F. N. Gygax, and A. Schenck (1995). Science 267, 1799.
B. Narymbetov, A. Omerzu, V. V. Kabanov, M. Tokumoto, H. Kobayashi, and D. Mihailovic (2000). Nature 407, 883.
T. L. Makarova, B. Sundqvist, R. Hohne, P. Esquinazi, Y. Kopelevich, P. Scharff, V. A. Davydov, L. S. Kashevarova, and A. V. Rakhmanina (2001). Nature 413, 716.
D. M. Guldi and N. Martin. In Fullerenes: From Synthesis to Optoelectronic Properties (Academic Publishers: Dordrech, The Netherlands, 2002).
K. M. Ho, A. A. Shvartsburg, B. Pan, Z. Y. Lu, C. Z. Wang, J. G. Wacker, J. L. Fye, and M. F. Jarrold (1998). Nature 392, 582.
A. J. Koblar, M. Horoi, I. Chaudhuri, T. Frauenheim, and A. A. Shvartsburg (2004). Phys. Rev. Lett. 93, 013401.
M. Menon and K. R. Subbasvramy (1994). Chem. Phys. Lett. 219, 219.
M. C. Piqueras, R. Crespo, E. Orti, and F. Tomas (1993). Chem. Phys. Lett. 213, 509.
S. Nagase and K. Kobayashi (1991). Chem. Phys. Lett. 187, 291.
X. G. Gong and Q. Q. Zheng (1995). Phys. Rev. B 52, 4756.
J. Zhao, L. Ma, and B. Wen (2007). J. Phys.: Condens. Matter 19, 226208.
C. Ray, M. Pellarin, J. L. Lermé, J. L. Vialle, M. Broyer, X. Blase, P. Mélinon, P. Kéghélian, and A. Perez (1998). Phys. Rev. Lett. 80, 5365.
M. Pellarin, C. Ray, J. Lermé, J. L. Vialle, M. Broyer, and P. Mélinon (2000). J. Chem. Phys. 112, 8436.
M. Ohara, Y. Nakamura, Y. Negishi, K. Miyajima, A. Nakajima, and K. Kaya (2002). J. Phys. Chem. A 106, 4498.
F. Tournus, B. Masenelli, P. Mélinon, X. Blase, A. Perez, M. Pellarin, M. Broyer, A. M. Flank, and P. Lagarde (2002). Phys. Rev. B 65, 165417.
Q. Sun, Q. Wang, P. Jena, B. K. Rao, and Y. Kawazoe (2003). Phys. Rev. Lett. 90, 135503.
S. M. Beck (1987). J. Chem. Phys. 87, 4233.
S. M. Beck (1989). J. Chem. Phys. 90, 6306.
H. Hiura, T. Miyazaki, and T. Kanayama (2001). Phys. Rev. Lett. 86, 1733.
K. Koyasu, M. Akutsu, M. Mitsui, and A. Nakajima (2005). J. Am. Chem. Soc. 127, 4998.
X. Huang, H. G. Xu, S. Lu, Y. Su, R. B. King, J. Zhao, and W. Zheng (2014). Nanoscale 6, 14617.
V. Kumar and Y. Kawazoe (2001). Phys. Rev. Lett. 87, 045503.
V. Kumar and Y. Kawazoe (2002). Phys. Rev. B 65, 073404.
V. Kumar and Y. Kawazoe (2003). Appl. Phys. Lett. 83, 2677.
H. Kawamura, V. Kumar, and Y. Kawazoe (2004). Phys. Rev. B 70, 245433.
H. Kawamura, V. Kumar, and Y. Kawazoe (2005). Phys. Rev. B 71, 075423.
J. Lu and S. Nagase (2003). Phys. Rev. Lett. 90, 115506.
G. Mpourmpakis, G. E. Froudakis, A. N. Andriotis, and M. Menon (2003). J. Chem. Phys. 119, 7498.
G. Mpourmpakis, G. E. Froudakis, A. N. Andriotis, and M. Menon (2003). Phys. Rev. B 68, 125407.
L. Ma, J. J. Zhao, J. G. Wang, Q. L. Lu, L. Z. Zhu, and G. H. Wang (2005). Chem. Phys. Lett. 411, 279.
L. Ma, J. J. Zhao, J. G. Wang, B. L. Wang, Q. L. Lu, and G. H. Wang (2006). Phys. Rev. B 73, 125439.
Z. U. Ren, F. Li, P. Guo, and J. G. Han (2005). J. Mol. Struct. 718, 165.
E. N. Koukaras, C. S. Garoufalis, and A. D. Zdetsis (2006). Phys. Rev. B 73, 235417.
J. Wang, Q. M. Ma, Z. Xie, Y. Liu, and Y. C. Li (2007). Phys. Rev. B 76, 035406.
J. Zhao, X. Huang, P. Jin, and Z. Chen (2015). Coordination Chemistry Reviews 289-290, 315.
C. Xiao, J. Blundell, F. Hagelberg, and W. A. Lester Jr. (2004). Int. J. Quantum Chem. 96, 416.
L. J. Guo, X. Liu, G. F. Zhao, and Y. H. Luo (2007). J. Chem. Phys. 126, 234704.
J. Wang and J. G. Han (2005). J. Chem. Phys. 123, 064306.
P. Guo, Z. Y. Ren, F. Wang, J. Bian, J. G. Han, and G. H. Wang (2004). J. Chem. Phys. 121, 12265.
J. G. Han and F. Hagelberg (2001). Chem. Phys. 263, 255.
J. G. Han and F. Hagelberg (2001). J. Mol. Struct. 549, 165.
J. G. Han, C. Xiao, and F. Hagelberg (2002). Struct. Chem. 13, 173.
J. G. Han, Z. Y. Ren, and B. Z. Lu (2004). J. Phys. Chem. A 108, 5100.
J. G. Han (2003). Chem. Phys. 286, 181.
J. Wang, Q. M. Ma, R. P. Xu, Y. Liu, and Y. C. Li (2009). Phys. Lett. A 373, 2869.
H. Hammer, J. K. Norskov (1995). Nature 376, 238.
R. F. Barrow, W. J. M. Gissane, and D. N. Travis (1964). Nature 201, 603.
C. Majumder, A. K. Kandalam, and P. Jena,(2006). Phys. Rev. B 74, 205437.
Q. Sun, Q. Wang, G. Chen, and P. Jena (2007). J. Chem. Phys. 127, 214706.
B. Kiran, X. Li, H. J. Zhai, L. F. Cui, and L. S. Wang (2004). Angew. Chem. Int. Ed. 43, 2125.
X. Li, B. Kiran, and L. S. Wang (2005). J. Phys. Chem. A 109, 4366.
C. Xiao, F. Hagelberg, and W. A. Lester Jr. (2002). Phys. Rev. B 66, 075425.
P. F. Zhang, J. G. Hana, and Q. R. Pu (2003). J. Mol. Struct. 635, 25.
F. C. Chuang, Y. Y. Hsieh, C. C. Hsu, and M. A. Albao (2007). J. Chem. Phys. 127, 144313.
J. Wang, Y. Liu, and Y. C. Li (2010). Phys. Lett. A 374, 2736.
H. Prinzbach, A. Weiler, P. Landenberger, F. Wahl, J. Wrth, L. T. Scott, M. Gelmont, D. Olevano, and B. Issendorff (2000). Nature 407, 60.
M. F. Jarrold (2000). Nature 407, 26.
C. Miller (1991). Science 252, 1092.
K. M. Ho, A .A. Shvartsburg, B. Pan, Z. Y. Lu, C. Z. Wang, J. G. Wacker, J. L. Fye, and M. F. Jarrold (1998). Nature 392, 582.
A. A. Shvartsburg, M. F. Jarrold, B. Liu, Z. Y. Lu, C. Z. Wang, and K. M. Ho (1998). Phys. Rev. Lett. 81, 4616.
B. X. Li and P. L. Cao (2000). Phys. Rev. A 62, 023201.
Q. Sun, Q. Wang, T. M. Briere, V. Kumar, and Y. Kawazoe (2002). Phys. Rev. B 65, 235417.
A. K. Singh, V. Kumar, and Y. Kawazoe (2005). Phys. Rev. B 71, 115429.
V. Kumar, A. K. Singh, and Y. Kawazoe (2006). Phys. Rev. B 74, 125411.
A. Grubisic, H. P. Wang, Y. J. Ko, and K. H. Bowena (2008). J. Chem. Phys. 129, 054302.
J. Wang and J. H. Liu (2009). J. Comput. Chem. 30, 1103.
J. Wang, Y. Liu, and Y. C. Li (2010). Phys. Chem. Chem. Phys. 12, 11428.
R. L. Zhou and B. C. Pan (2007). Phys. Lett. A 368, 396.
J. Bai, L. F. Cui, J. L. Wang, S. Yoo, X. Li, J. Jellinek, C. Koehler, Th. Frauenheim, L. S. Wang, and X. C. Zeng (2006). J. Phys. Chem. A 110, 908.
J. Bai and X. C. Zeng (2007). Nano 2, 109.
S. Yoo, J. J. Zhao, J. L. Wang, and X. C. Zeng (2004). J. Am. Chem. Soc. 126, 13845.
S. Yoo, N. Shao, C. Koehler, T. Fraunhaum, and X. C. Zeng (2006). J. Chem. Phys. 124, 164311.
J. Li, J. Wang, H. Y. Zhao, and Y. Liu (2013). J. Phys. Chem. C 117, 10764.
Acknowledgments
The authors thank Dr. N. E. Davison for his help with the language. This work is supported by the National Natural Science Foundation of China (Grant Nos. 11274089, U1331116 and 11304076), the Natural Science Foundation of Hebei Province (Grant Nos. A2012205066 and A2015205179), the Science Foundation of Hebei Education Award for Distinguished Young Scholars (Grant No. YQ2013008), and the Program for High-level Talents of Hebei Province (Grant No. A201500118). We also acknowledge partial financial support from the 973 Project in China under Grant No. 2011CB606401.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, J., Liu, Y. Magnetic Silicon Fullerenes: Experimental Exploration and Theoretical Insight. J Clust Sci 27, 861–873 (2016). https://doi.org/10.1007/s10876-015-0959-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-015-0959-6