
Abstract
Background
Chronic Granulomatous Disease (CGD) is a rare immunodeficiency disorder characterized by impaired phagocytic function, leading to recurrent infections and granuloma formation. Genetic mutations in NADPH oxidase complex components, such as CYBB, NCF1, NCF2, and CYBA genes, contribute to the pathogenesis. This case report explores the possible ocular and hematologic complications associated with CGD.
Case Presentation
A 6-year-old girl with a history of vitrectomy, membranotomy, and laser therapy due to congenital blindness (diagnosed with chorioretinopathy) was referred to the hospital with generalized ecchymosis and thrombocytopenia. Diagnostic workup initially suggested chronic immune thrombocytopenic purpura (ITP). Subsequent admissions revealed necrotic wounds, urinary tract infections, and recurrent thrombocytopenia. Suspecting immunodeficiency, tests for CGD, Nitroblue tetrazolium (NBT) and dihydrorhodamine (DHR) were performed. She had a low DHR (6.7), and her NBT test was negative (0.0%). Her whole exome sequencing results confirmed autosomal recessive CGD with a homozygous NCF1 mutation.
Conclusion
This case underscores the diverse clinical manifestations of CGD, including recurrent thrombocytopenia and possible early-onset ocular involvement. The diagnostic challenges highlight the importance of a multidisciplinary approach involving hematologists, immunologists, and ophthalmologists for accurate diagnosis and management. The rare coexistence of ITP in CGD emphasizes the intricate link between immunodeficiency and autoimmunity, requiring tailored therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Chronic Granulomatous Disease (CGD) is a rare inherited immunodeficiency disorder characterized by impaired phagocytic function, rendering affected individuals susceptible to recurrent and severe bacterial and fungal infections and granuloma formation. The global prevalence of CGD is estimated to be around 1 in 200,000 to 1 in 250,000 live births, underscoring its rarity in the spectrum of genetic disorders [1]. CGD is typically inherited in an X-linked or autosomal recessive manner and results from mutations in genes encoding components of the phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex. The most prevalent genetic mutations involve the CYBB gene (X-linked) and the autosomal genes NCF1, NCF2, and CYBA [2].
The pathophysiology of CGD revolves around the impaired function of the phagocytic NADPH oxidase enzyme complex, resulting in the inability of phagocytes, such as neutrophils, monocytes, and macrophages, to generate reactive oxygen species essential for microbial killing. This defective immune response predisposes individuals with CGD to persistent infections and dysregulated inflammatory responses, contributing to the diverse clinical spectrum observed in affected patients [2, 3].
Most patients typically encounter their initial CGD symptoms within the first year of life. In CGD patients, the most affected sites include the lung, lymph nodes, liver, and skin [4]. However, other complications, such as urinary tract infections, immune thrombocytopenia, juvenile idiopathic arthritis, photosensitivity, gingivitis, etc., have also been present in CGD patients [2, 3, 5, 6]. Ocular manifestations, such as chorioretinal lesions, uveitis, keratitis, and optic nerve disease, which are not common, were also reported in some CGD cases [7].
Here, we report a case that presented with thrombocytopenia and had an early-onset ocular involvement that could be possibly associated with CGD, highlighting the importance of a multidisciplinary approach to this disease.
Case Presentation
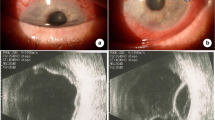
A 6-year-old girl, born to a consanguineous family, with a history of recent vitrectomy, membranotomy, and laser therapy due to rhegmatogenous retinal detachment (RRD) and chorioretinopathy (presented with congenital left eye blindness and severe vision loss in her right eye) was referred to the Children Medical Center Hospital, Tehran, Iran, in 2016. The patient’s chief complaint was generalized ecchymosis, and she was admitted to the hospital due to thrombocytopenia (platelet: 6000/µL). There was no history of similar ocular and hematologic manifestations in her parents and siblings (2 brothers and 1 sister). To determine the cause of her thrombocytopenia, she underwent diagnostic testing for potential diseases that could be causing this condition. Her laboratory workup for systemic lupus erythematosus, including C3, C4, CH50, anti-double-stranded DNA (anti-dsDNA) antibody, antinuclear antibody (ANA), perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA), and cytoplasmic ANCA (c-ANCA) was normal. The results of her flow cytometry on bone marrow aspiration/biopsy sample (conducted to rule out hematologic malignancies) and abdominal ultrasound were normal. In eye examination, retinal and optic nerve atrophy was detected in both eyes. Her TORCH test by Real-time polymerase chain reaction (PCR), blood culture, and urine culture were negative. During hospitalization, she received intravenous immunoglobulin (IVIG) and was discharged from the hospital with a platelet number of 159,000/µL. Based on her frequent hospitalizations for thrombocytopenia, she was diagnosed with chronic immune thrombocytopenic purpura (ITP) and received corticosteroids intermittently.
At the age of 9, she was admitted to the hospital because of a necrotic wound on the posterior side of her right thigh, which first appeared as cellulitis two months ago and did not respond to oral antibiotic therapy. Her soft tissue ultrasonography showed two sites with no vascularity (size: 8*20 mm and 5*19 mm) and localized hyperemia under her wound on the posterior side of her right thigh. During her hospitalization, the size of the wound decreased with intravenous antibiotic therapy. Her Tuberculin Skin Test and interferon-gamma release assays (IGRA) were also negative. At the age of 10, she was admitted to the hospital due to a urinary tract infection and a high serum creatinine (2.4 mg/dL). Her kidney, urinary tract, and bladder ultrasounds were normal. After hydration and intravenous antibiotic therapy (ceftriaxone) for two days, she was discharged from the hospital with a serum creatinine of 0.7 mg/dL. She experienced a urinary tract infection (Escherichia coli) 3 months later and was treated with oral antibiotic therapy. Her further screening for metabolic diseases was also negative.
At the age of 13, she was hospitalized due to thrombocytopenia (platelet: 11,000/µL), presenting with an ecchymosis with a size of 2*4 cm. Due to the history of recurrent infections, the patient was suspected of immunodeficiency disorders. Nitroblue tetrazolium (NBT) and dihydrorhodamine (DHR) tests were performed to assess whether the patient had CGD. She had a low DHR (6.7), and her NBT test was negative (0.0%) (Fig. 1). Bone marrow biopsy and aspiration were repeated, which was normal for the second time. Whole Exome Sequencing (WES) was performed, showing a homozygous genotype for mutated Neutrophil Cytosolic Factor 1 (NCF1) ENST00000289473.4 (NM_000265): c.73_74delGT, p.Y26Hfs*26. Other possible assessed candidates, including COL2A1, TCF4, ACO2, and LRIT3, could not explain the clinical history of the patient (Table 1). Based on the results of the aforementioned tests, our patient was diagnosed with autosomal recessive CGD. She was given Romiplostim for six weeks, but her platelet count did not increase. Consequently, IVIG was administered, raising her platelet count to 80,000. After being discharged, she was referred for a bone marrow transplantation.

DHR histogram. The histograms above show neutrophil NADPH oxidase activity of the case and a healthy control in both an unstimulated sample and a sample stimulated with phorbol myristate (PMA). The result is consistent with CGD
Discussion
This case report details the complex medical history of a girl with congenital left eye blindness and severe vision loss in her right eye, who subsequently presented with recurrent thrombocytopenia and skin and urinary tract infections. The initial diagnosis of chronic ITP was made based on her frequent hospitalizations for thrombocytopenia. However, her other clinical manifestations raised concerns about the possibility of an underlying immunodeficiency disorder. The suspicion of immunodeficiency disorders was confirmed through specific tests for CGD. The patient’s low DHR test result and a negative NBT test, along with a homozygous genotype for mutated NCF1 identified through WES, established the diagnosis of autosomal recessive CGD.
CGD originates from mutations that lead to the loss or functional inactivation of subunits within the NADPH oxidase complex. The NADPH oxidase complex comprises five main components, and mutations in genes encoding gp91phox, p22phox, p47phox, p67phox, or p40phox contribute to the formation of the NADPH oxidase complex, playing a significant role in various CGD phenotypes. Also, mutations in CYBC1 (EROS), the stabilizer of the NADPH oxidase complex, can lead to CGD5 [8]. The majority of CGD mutations follow an autosomal recessive inheritance pattern; however, the CYBB variant is distinctive in being inherited as X-linked recessive [3, 9]. Neutrophilic granulocytes are commonly employed to assess the activation of NADPH oxidase in these cells. NADPH oxidase assays, such as the cytochrome c reduction assay and the NBT slide assay, measure superoxide production. Hydrogen peroxide levels can be determined by the Amplex Red assay, and the DHR assay can be used to assess NADPH oxidase activity. While the patient’s clinical history may offer insights into the genetic inheritance pattern, genetic testing remains instrumental in identifying specific genetic mutations associated with CGD [10].
There is a possibility that the ocular manifestation of our patient is associated with CGD. The initial recognition of ocular manifestations in CGD dates back to 1965, with chorioretinitis being the first documented presentation. Subsequently, numerous authors have documented a spectrum of ocular signs associated with the disease, with the most prevalent including chorioretinal lesions, uveitis, keratitis, conjunctivitis, corneal ulcers, and optic nerve atrophy [11,12,13,14]. Histopathological reports of ocular lesions related to CGD are scarce and have not shown evidence of local infectious processes [12, 14]. Chorioretinal lesions are thought to be secondary to granuloma development or sterile inflammation due to activated abnormal phagocytic cells [15]. The formation of granulomas could be attributed to the abnormal functions of phagocytic cells in the retinal pigment epithelium [12].
Various hypotheses can be posited regarding the origin of these lesions. The pigment epithelium, whose properties can be likened to those of macrophages, may contribute to the formation of these scars. Pigment epithelium cells, through NADPH oxidase, generate oxygen derivatives that, in cases of CGD, might induce granuloma development due to an impaired ability to digest cellular debris from cones and rods, thereby initiating an inflammatory cascade [16]. According to Valluri et al. [11], endocular granulomas could arise in the presence of a sufficient quantity of bacterial or fungal antigens. These sterile abscesses result from an inappropriate inflammatory response, persisting long after the infectious process has been sterilized. The disposition and morphology of these lesions resemble “septic metastases,” yet no microorganisms are identified within them [16].
While immunodeficiency and autoimmunity are often perceived as opposing facets of the immune response, they are intricately linked. ITP in CGD represents a unique and challenging aspect of the disease’s clinical spectrum. This rare coexistence of ITP and CGD has been previously reported in several case reports [17, 18]. Abnormalities in T-cell function and cytokine production have been reported in CGD, and these immune dysregulations may contribute to the development of autoimmune complications. Also, cross-reactive auto-antibodies that are produced during impaired phagocytosis could also explain autoimmune disorders in CGD patients [17, 18]. Rituximab and cyclosporine have shown promising results in treating ITP in CGD cases [17]. There is a report of a case of autosomal recessive CGD (NCF1 mutation) with IgA deficiency and refractory autoimmune thrombocytopenia that showed an excellent response to Rituximab [19].
Based on the clinical manifestations of our case, a holistic approach, considering the immunodeficiency, genetic factors, and potential interactions with other CGD-related manifestations, is essential for effective diagnosis and management. Collaborative efforts between hematologists, immunologists, and infectious disease specialists are crucial in providing comprehensive care for individuals with CGD. The article also emphasizes the role of ophthalmologists in the multidisciplinary management of CGD, underscoring the need for early identification and intervention for possible ocular complications in these patients. It should be noted that further research is required to confirm that the ocular complications mentioned in this case report were purely due to CGD.
Data Availability
All data associated with this study is included in this published article.
Code Availability
Not applicable.
References
Lee E, Oh SH, Kwon JW, Kim BJ, Yu J, Park CJ, et al. A case report of chronic granulomatous disease presenting with aspergillus pneumonia in a 2-month old girl. Korean J Pediatr. 2010;53(6):722–6.
Rider NL, Jameson MB, Creech CB. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and genetic basis of Disease. J Pediatr Infect Dis Soc. 2018;7(suppl1):S2–5.
Arnold DE, Heimall JR. A review of chronic Granulomatous Disease. Adv Ther. 2017;34(12):2543–57.
Miladinovic M, Wittekindt B, Fischer S, Gradhand E, Kunzmann S, Zimmermann SY, et al. Case Report: symptomatic chronic Granulomatous Disease in the Newborn. Front Immunol. 2021;12:663883.
Dar-Odeh NS, Hayajneh WA, Abu-Hammad OA, Hammad HM, Al-Wahadneh AM, Bulos NK, et al. Orofacial findings in chronic granulomatous disease: report of twelve patients and review of the literature. BMC Res Notes. 2010;3:37.
Sadrosadat T, Ziaee V, Aghighi Y, Moradinejad MH, Movahedi M. Presence of a Juvenile Idiopathic Arthritis and Chronic Granulomatous Disease in a child. Iran J Pediatr. 2015;25(2):e365.
Khetarpal S, Yeh S, Smith JA. Ophthalmic manifestations of Chronic Granulomatous Disease. Investig Ophthalmol Vis Sci. 2008;49(13):809.
Thomas DC, Charbonnier L-M, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. 2019;143(2):782–e51.
Lent-Schochet D, Jialal I. Chronic Granulomatous Disease. StatPearls. Treasure Island (FL) with ineligible companies. Disclosure: Ishwarlal Jialal declares no relevant financial relationships with ineligible companies.: StatPearls Publishing Copyright © 2024. StatPearls Publishing LLC.; 2024.
Kulkarni M, Hule G, de Boer M, van Leeuwen K, Kambli P, Aluri J, et al. Approach to Molecular diagnosis of chronic granulomatous disease (CGD): an experience from a large cohort of 90 Indian patients. J Clin Immunol. 2018;38(8):898–916.
Valluri S, Chu FC, Smith ME. Ocular pathologic findings of chronic granulomatous disease of childhood. Am J Ophthalmol. 1995;120(1):120–3.
Goldblatt D, Butcher J, Thrasher AJ, Russell-Eggitt I. Chorioretinal lesions in patients and carriers of chronic granulomatous disease. J Pediatr. 1999;134(6):780–3.
Djalilian AR, Smith JA, Walsh TJ, Malech HL, Robinson MR. Keratitis caused by Candida Glabrata in a patient with chronic granulomatous disease. Am J Ophthalmol. 2001;132(5):782–3.
Al-Muhsen S, Al-Hemidan A, Al-Shehri A, Al-Harbi A, Al-Ghonaium A, Al-Saud B, et al. Ocular manifestations in chronic granulomatous disease in Saudi Arabia. J Aapos. 2009;13(4):396–9.
Buggage RR, Bauer RM 2nd, Holland SM, Santos CI, Chan CC. Uveitis and a subretinal mass in a patient with chronic granulomatous disease. Br J Ophthalmol. 2006;90(4):514–5.
Locatelli A, Béné MC, Zuily S, Angioi-Duprez K. [Ocular manifestations in chronic granulomatous disease]. J Fr Ophtalmol. 2013;36(9):789–95.
Treliński J, Chojnowski K, Kurenko-Deptuch M, Kasznicki M, Bernatowska E, Robak T. Successful treatment of refractory autoimmune thrombocytopenia with rituximab and cyclosporin A in a patient with chronic granulomatous disease. Ann Hematol. 2005;84(12):835–6.
Sarkar S, Mondal DR, Nandi M. Chronic granulomatous disease presenting as immune thrombocytopenic purpura. Sri Lanka Journal of Child Health. 2014;2014; 43: 177–179:177 – 79.
Shamsian B, Mansouri D, Pourpak Z, Rezaei N, Chavoshzadeh Z, Jadali F, et al. Autosomal recessive chronic Granulomatous Disease, IgA Deficiency and Refractory Autoimmune Thrombocytopenia responding to Anti-CD20 monoclonal antibody. Iran J Allergy Asthma Immunol. 2008;7:181–4.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
Conceptualization: FK; Formal analysis and investigation: SK, NR, FK, MD; Writing - original draft preparation: SK; Writing - review and editing: NR, FK, SK; Imaging studies and patient examination: FK, MD; Supervision: FK. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study is approved by the ethics committee at the Tehran University of Medical Sciences (Approval ID: IR.TUMS.CHMC.REC.1401.179).
Consent to Participate
Informed consent was obtained from the patient’s father to participate in the study.
Consent for publication
Informed consent was obtained from the patient’s father for publication of this case report and any accompanying data.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khanmohammadi, S., Rezaei, N., Kompani, F. et al. Immune Thrombocytopenic Purpura (ITP) and Chorioretinopathy in Chronic Granulomatous Disease: A Case Report. J Clin Immunol 44, 125 (2024). https://doi.org/10.1007/s10875-024-01731-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10875-024-01731-8