
Abstract
Purpose
Major histocompatibility complex class II (MHC-II) deficiency is a rare inborn error of immunity (IEI). Impaired antigen presentation to CD4 + T cells results in combined immunodeficiency (CID). Patients typically present with severe respiratory and gastrointestinal tract infections at early ages. Hematopoietic stem cell transplantation (HSCT) is the only curative therapy.
Methods
We describe the clinical, immunologic, and genetic features of eighteen unrelated Iranian patients with MHC-II deficiency.
Results
Consanguinity was present in all affected families. The median age at the initial presentation was 5.5 months (range 7 days to 18 years). The main symptoms included failure to thrive, persistent diarrhea, and pneumonia. Autoimmune and neurologic features were also documented in about one-third of the patients, respectively. Thirteen patients carried RFXANK gene mutations, two carried RFX5 gene mutations, and three carried a RFXAP gene mutation. Six patients shared the same RFXANK founder mutation (c.162delG); limited to the Iranian population and dated to approximately 1296 years ago. Four of the patients underwent HSCT; three of them are alive. On the other hand, nine of the fourteen patients who did not undergo HSCT had a poor prognosis and died.
Conclusion
MHC-II deficiency is not rare in Iran, with a high rate of consanguinity. It should be considered in the differential diagnosis of CID at any age. With the limited access to HSCT and its variable results in MHC-II deficiency, implementing genetic counseling and family planning for the affected families are mandatory. We are better determined to study the c.162delG RFXANK heterozygous mutation frequency in the Iranian population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major histocompatibility complex class II (MHC-II) molecules are transmembrane glycoproteins that specialized for the presentation of peptides to the T cell receptor (TCR); thus, they have key roles in the maturation of CD4 + T lymphocytes in the thymus as well as CD4 + T cell–dependent immune responses in the periphery [1,2,3]. Three different human MHC-II isotypes (HLA-DR, HLA-DP, and HLA-DQ) are expressed over antigen-presenting cells (APCs) such as dendritic cells, macrophages, and B lymphocytes [3, 4]. MHC-II deficiency is a rare inborn error of immunity (IEI) first described in the 1980s [5,6,7,8]. It is defined by the lack of MHC-II expression on APC cells and the absence of CD4 + T cell–dependent immune responses. Moreover, perturbation of central and peripheral immune tolerance mechanisms, specifically the defects in the maturation and function of thymic epithelial cells (TEC), underlies immune dysregulation in MHC-II deficiency [9]. The disease is caused by mutations in four distinct genes (CIITA, RFXANK, RFXAP, and RFX5) encoding trans-acting regulatory factors required to transcribe MHC-II genes and MHC-II expression [3, 4, 10, 11].
The resulting combined immunodeficiency (CID) leads to severe and recurrent respiratory and gastrointestinal infections. In most patients, infections start within the first year of life, but some have milder courses of disease and are diagnosed later, at ages of up to 15 years [12, 13]. Laboratory study typically shows CD4 + lymphocytopenia in the presence of normal total T cell numbers. Absence or very low HLA-DR expression on immune cells and abnormal antigen-specific cellular and humoral responses are also seen [4, 14]. MHC-II deficiency has a dismal outcome. However, allogeneic hematopoietic stem cell transplantation (HSCT) is the only available curative treatment with variable success.
MHC-II deficiency is rare; most patients are reported from North Africa [15,16,17,18,19,20]. The remaining patients are occasionally reported from other nationalities [21,22,23,24,25]. We present the clinical, immunological, and genetic features of eighteen unrelated Iranian patients with MHC-II deficiency. Six of them have the same RFXANK mutation, and we investigated and proved the possibility of a founder effect.
Materials and Methods
We report on 18 unrelated Iranian MHC-II-deficient patients in this case series. Thirteen patients are new, and five have been reported previously [26,27,28,29,30]. Informed consent for participating in the study was obtained from the patients or their parents. The Children’s Medical Center Institutional Ethics Committee, affiliated with the Tehran University of Medical Sciences, approved the study. We thoroughly reviewed patient medical records and collected detailed demographic and clinical data: gender, age at presentation, diagnosis, family history, clinical presentation, diagnostic workup, management, and outcome. Clinical data from previously published cases have been compiled from the pertinent articles.
Clinical whole exome sequencing (WES) was performed on patient samples, as reported previously [31]. Exome capture was performed with the Sure Select Human All Exon 50 Mb kit (Agilent Technologies). Paired-end sequencing was performed on a HiSeq 2000 (Illumina), generating 100-base reads. Bi-directional sequence reads were assembled and aligned to the human genome build GRCh38/hg19.
Downstream processing and variant calling were performed with the genome analysis toolkit, SAMtools, and Picard. Substitution and InDel calls were made with GATK Unified Genotyper. All variants were annotated using an annotation software system that was developed in-house. All the variants have been documented by Sanger sequencing.
Common haplotype analysis on RFXANK variant (c.126delG) was conducted to prove the founder effect of c.126delG in Iranian patients. Haplotype analysis was determined by identifying shared regions of homozygous variants from each affected individual with the homozygous c.126delG variant at chr19: 191,941,08.
Results
Epidemiologic Features
We investigated eighteen patients from unrelated families with MHC-II deficiency with varying clinical and immunological phenotypes. All the patients were of Iranian origin, born to consanguineous families. The median age at the initial presentation was 5.5 months (range 7 days to 18 years). The median age at immunodeficiency diagnosis and the genetic confirmation was 12 months and 25 months, respectively. Six of the 18 patients had another sibling who had died with similar symptoms.
Clinical Characteristics
The patients’ clinical features are summarized in Table 1 (the detailed clinical data is presented in the supplementary Table S1).
All the patients received attenuated oral poliovirus vaccine (OPV) and Bacillus Calmette–Guerin (BCG) at birth. All the patients suffered from several manifestations before they came to our attention. Failure to thrive (FTT) was present in fifteen (83.3%) patients.
Respiratory Tract Infections
Recurrent pneumonia was reported in 14 patients (77.7%) and occurred before 1 year of age in 10 patients (55.5%). Three patients (P2, P5, and P15) developed COVID-19 pneumonia after SARS-CoV-2. P2 developed cardiomyopathy during COVID-19, which was finally resolved. P5 died after COVID-19 complications. Bronchoalveolar lavage (BAL) was positive for CMV infection in P6 and Acinetobacter spp. in P5.
Six patients (33.33%) had recurrent acute otitis media that four of which began before the age of 2 years and required a trans-tympanic drain in P9. Chronic sinusitis was documented in 2 patients (P10, P11). P1 died from sinus mucormycosis with brain invasion.
Gastrointestinal Manifestations
Fourteen patients (77.7%) had gastrointestinal manifestations. Persistent diarrhea (66.7%) was a common gastrointestinal problem. A histologic study of intestinal mucosa was performed in six patients. This study revealed autoimmune enteropathy in one patient (P2), nodular lymphoid hyperplasia in two (P11, P17), and granulation tissue in the colon in another (P5) patient. Seven patients (38.8%) displayed chronic oral candidiasis complicated by esophageal involvement in P11. Four patients were diagnosed with CMV colitis (P1, P2, P5, and P8); two of whom (P1, P2) developed bowel perforation. Four patients (P1, P2, P8, and P11) had recurrent diarrhea caused by C. parvum. In P11, cryptosporidium infection resulted in sclerosing cholangitis and hepatorenal syndrome. Recurrent diarrhea caused by norovirus was observed in one patient (P2) and persisted until death. Hepatic abnormalities were not frequent, and three patients had hepatomegaly and/or high serum levels of liver transaminases (P6, P8, and P11).
Other Infections
Two patients (P7, P17) had BCG lymphadenitis that resolved without complication.
Blood cultures were positive for Pseudomonas aeruginosa (P2) and Enterobacter cloacae (P5). P4 had CMV viremia without definitive organ involvement, and P13 had CMV viremia with brain involvement. One patient (P10) had recurrent UTI caused by Klebsiella pneumonia.
P18 developed vaccine-associated paralytic poliomyelitis (VAPP) at 7 months. He excreted vaccine-derived poliovirus type 2 (VDPV2).
Other Manifestations
Allergy was observed in 50% of the patients. Six patients had asthma, and four (P5, P8, P9, and P16) had atopic dermatitis. An allergic cutaneous drug reaction was observed in two patients (P6 after fluconazole and P8 after cefixime).
Autoimmune hemolytic anemia (AIHA) occurred in three patients (P1, P3, and P10) at ages 7, 2.5, and 36 years, respectively. P1 and P10 were treated with oral prednisolone and azathioprine, and P3 was treated with prednisolone and cyclosporine. Two other patients (P14, P4) had peripheral neutropenia with no detectable autoantibodies. Two patients (P8, P17) had a clinical diagnosis of juvenile idiopathic arthritis (JIA).
Neurologic complications unrelated to poliovirus occurred in seven patients. Neurodevelopmental delay (NDD) and hypotonia were observed in two patients (P12 and P16) with normal brain imaging. In addition to NDD, lower limb spasticity and axial hypotonia during infancy were observed in P13. Brain computed tomography (CT) scan revealed thalamus and basal ganglia calcification, probably caused by CMV infection. P14 patient had ataxic gait and lower limb muscle weakness at 2 years of age with normal brain magnetic resonance imaging (MRI). P1 presented with chorea athetosis at age 10 years. Brain imaging showed brain atrophy and mild ventriculomegaly. P7 experienced encephalopathy without documented infection; his symptoms improved after antibiotics, corticosteroid, and IVIG treatment. P15 had dysmorphic features, mild hypotonia, and brain atrophy in brain MRI. There were no enteroviruses or HSV found in the cerebrospinal fluid of any of the six patients who had neurological problems.
Immunologic Findings
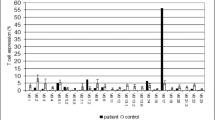
All patients displayed a total absence of MHC class II (HLA-DR) molecule expression on B cells except two patients (P13, P17). There were low absolute CD4 + T cell counts in 13 of the 18 patients (72.2%). CD4 + T cell counts were below 500 cells/μL for ten patients (55.5%) between the ages of 4 months and 32 years. CD8 + T cell counts were low in three patients (16.6%) and high in another six (33.3%). All the patients, even those with normal T cell CD4 + count, had a low CD4:CD8 ratio.
Natural killer cell (CD3–CD16 + /CD56 +) counts were low in 3 of the 18 (16.6%) patients tested. In vitro proliferative responses of T cells to mitogen phytohemagglutinin (PHA) were normal for all patients, but no proliferation in response to antigens or delayed type hypersensitivity (DTH) to tetanus toxoid was observed in 11 of 12 tested patients. B cell immunity was also profoundly impaired. Twelve of the 18 patients studied (66.6%) had normal numbers of circulating B lymphocytes, whereas the other 6 (33.3%) had low circulating B lymphocytes. Before the initiation of IgG substitution therapy, 12 of 16 patients (75%) had low levels of IgG, nine had low levels of IgA (56.3%), and 10 of 15 had low levels of IgM (66.6%). Two patients (P5, P17) had high baseline IgG levels at 2 and 3.5 years, respectively.
No specific antibodies against pneumococcal or protein vaccines could be detected in the 7 of 11 (63.6%) patients tested. Immunologic data are summarized in Table 2 (the detailed immunologic data is presented in the supplementary Table S2).
Genetic Results
We performed WES for the fourteen patients. Sanger sequencing confirmed the variants. All the parents were heterozygous.
Seven different mutations of the RFXANK gene have been characterized in thirteen unrelated patients (Table 3, Fig. 1a). The RFXANK variants are compared to the homozygous coding missense and predicted loss-of-function RFXANK mutations taken from GnomAD (Fig. 1b). Our reported variants are private and have combined annotation-dependent depletion (CADD) scores above the mutation significance cutoff (MSC) of 21.4 [32].

MHC-II deficiency in the 18 Iranian patients. a Schematic representation of the RFXANK protein. ARD, ankyrin-repeat domain; PEST, activation domain rich in acidic amino acids. b Population genetics of RFXANK. The minor allele frequency (MAF) and CADD scores for all non-synonymous variants reported in the gnomAD database are shown. Seven homozygous variants found in our cohort are also shown. c Schematic representation of the RFXAP protein. NLS, nuclear localization signal. d Schematic representation of the RFX5 protein. NTD, N-terminal domain; DBD, DNA-binding domain; P, PxLPxI/L motif. The previously reported variants are indicated in black, while those of our patients are indicated in red
These include a deletion leading to a frameshift (P1–P6), three nonsense mutations (P9, P12, P13), one missense mutation (P8), and two splice-site mutations (P7, P10, P11). All the mutations affect the integrity of the ankyrin repeat domain (ARD), which is essential for the function of RFXANK [33, 34].
The c.162delG is a frameshift deletion documented in six unrelated patients. This mutation leads to an early termination codon, leaving a truncated protein. This is a novel mutation not reported in other populations and seems limited to the Iranian population.
The occurrence of homozygosity for the c.126delG mutation in these six kindreds of Iranian origin strongly suggested a founder effect. An analysis of the WES data showed that the index cases homozygous for the c.126delG mutation share a common homozygous haplotype around RFXANK, encompassing 2.7 Mbp corresponding to 83 SNVs (Fig. 2). The ESTIAGE method [35] estimated the age of the most recent common ancestor (MRCA) of the six patients at 48 generations [95% confidence interval (26–91)]. Assuming a generation time of 27 years [36], the MRCA of these patients with the c.126delG mutation would have lived about 1296 (702–2457) years ago.

Shared haplotype around the RFXANK c.162delG mutation for the six carriers. The distance from the mutation is represented on the Y-axis. Long continuous stretches of homozygosity were observed around the gene, consistent with its recessive mode of inheritance, and haplotypes were unambiguously derived from genotypes. The dbsnp reference numbers of the first (top) and last (bottom) unambiguous variants within the haplotype are reported for each carrier
The missense mutation p.Asp121His has been reported previously, resulting in the loss of the RFXANK function [24]. Two novel splice-site mutations are reported here. Splice-site mutation c.438 + 5G > A documented in two patients (P10, P11) seems responsible for a milder form of the disease. Three different RFXAP gene mutations have been identified (Table 3, Fig. 1c). These include two frameshift mutations (P15, P16) resulting from deletion and a nonsense mutation (P14). All these mutations lead to the synthesis of truncated proteins lacking the C-terminal glutamine-rich domain, which is known to be necessary for the RFX complex formation [37]. Two mutations of the RFX5 gene have been identified (Table 3, Fig. 1d). These include a missense (P17) and a nucleotide duplication (P18). The missense mutation lies in the DNA-binding domain (DBD) and is novel. The DBD of RFX5 is essential for association with RFXANK and RFXAP, correct assembly of the RFX complex in solution, and specific binding to the XX-box target site [38, 39]. The duplication mutation is an insertion leading to several amino acid changes, modifying the protein structure.
Outcome and Treatment
HSCT
In our series, four (P3, P6, P14, and P16) of the 18 patients (23.5%) underwent HSCT (Fig. 3). The indications for HSCT are mostly related to clinical status (age and infection state). The median age at HSCT was 1.5 years (range 1–5 years). Three of the four patients who received HSCT (75%) are alive. All the patients received reduced intensity conditioning (RIC). The RIC regimen consisted of a combination of intravenous (IV) fludarabine (30 mg/m2) administered for five consecutive days (days 8 to 4), IV melphalan (70 mg/m2) for two consecutive days (days 3 and 2), and IV horse anti-thymocyte globulin (10 mg/kg) for four consecutive days (days 4 to 1). IV cyclosporine A (CSA) (1.5 mg/kg daily is used on day 1; then 3 mg/kg from day + 7 in PBSC to day + 11 in BMSC) and IV methylprednisolone (1 mg/kg/day (day + 5 to + 7), then 0.5 mg/kg/day by day + 14) are used as GVHD prophylaxis.

Five-year overall survival curves (Kaplan–Meier) for patients who underwent HSCT and those who did not undergo HSCT
The patients received grafts from a matched-related donor (MRD, P16), HLA-identical sibling (P3), one-locus mismatched-related donor (MMRD, P14), and unrelated full-matched donor (P6). Primary engraftment occurred in all patients, but two experienced secondary graft failure (P6, P16); P6 underwent the second HSCT from the same donor, and she died after 1 month due to transplant-related complications (sepsis). P16 is alive, but re-transplantation is challenging because of her severe neurologic impairment. P3 and P14 developed full donor chimerism and doing well clinically.
The Natural History and Outcome of Patients Who Did Not Undergo HSCT
Fourteen patients did not undergo HSCT (Table S3, Fig. 3). The clinical course was unfavorable in these patients, with the progression of infectious complications to death in nine patients (64.2%; range: 11 months to 15 years; mean age at death: 33 months). P1 died from severe mucormycosis infection. P2 died from peritonitis due to terminal ileum perforation after a norovirus infection. P11 had a clinical course like other patients in early childhood but displayed milder symptoms. The frequency of infection in this patient increased with age. Immunologic investigations revealed no significant differences from other patients. He died at 15 years of age because of hepatorenal failure due to cryptosporidiosis.
P12 died from severe pneumonia. P18 had a severe progressive respiratory failure due to VAPP. Other patients (P7, P13, and P15) died from undefined systemic inflammatory response syndrome (SIRS).
Five patients who did not undergo HSCT (35.7%) are still alive at the time of this report at a median age of 10 years (range: 6–42 years).
P4 has had recurrent diarrhea and chronic bronchopneumonia since age four and was diagnosed at 4.5. He is now 9 years old.
P8 presented with chronic diarrhea, upper respiratory infections, and oral candidiasis since early infancy and was diagnosed with MHC-II deficiency at the age of 2 years. She had recurrent diarrhea caused by C. parvum and CMV colitis during follow-up. She developed JIA for 8 years. He is now 10 years old.
P9 has had symptoms since the 6 months of life and was diagnosed with MHC-II deficiency at 9 months. She presented recurrent upper and lower respiratory infections (otitis media with effusion leading to ventilation tube insertion) and persistent diarrhea. She is 6 years old and has FTT, short stature, and hypothyroidism.
P10 has had symptoms since the age of 18 years and was diagnosed with MHC-II deficiency at the age of 37 years. Her symptoms worsen after the age of 35 years, with an increase in the frequency and severity of infections. She had AIHA and positive antineutrophil cytoplasmic antibodies (ANCA), requiring azathioprine treatment at the age of 36 years. She also has recurrent UTI (K. pneumonia), recurrent upper respiratory infections, and chronic bronchopneumonia. She is still alive on supportive care (IgG treatment and antibiotic prophylaxis with trimethoprim-sulfamethoxazole). P17 presented with recurrent pneumonia since 2 months and was diagnosed at 12 months. At the age of 2 years, he presented with JIA. He is 12 years old.
Discussion
We describe the clinical, immunologic, and genetic features of eighteen unrelated patients with MHC-II deficiency from Iran. In most of our patients, clinical signs and outcomes were similar to those reported in other groups of patients [4, 15, 16, 22, 23, 40].
The clinical features mainly included FTT and bacterial and severe viral infections in the respiratory and gastrointestinal systems. Viral infections are especially associated with poor outcomes, with patients dying during childhood [22, 41]. In our series, poliovirus, SARS-CoV-2, and norovirus infections are also associated with poor prognosis. One of our patients developed VAPP after OPV inoculation at birth. It is better to monitor these patients for virus shedding in the stool at the time of diagnosis and regularly after that [42, 43]. Chronic infection with C. parvum is associated with sclerosing cholangitis in some patients [15, 22]. Indeed, hepatic involvement appears to be associated with early mortality [15].
Allergy was observed in 50% of our patients. The prevalence of allergies in our patients was higher than in the general population (10–12%) or even other CIDs (20%) in general [44, 45]. MHC-II deficiency might be implicated in driving allergic immune dysregulation. The presence of autoimmunity in one-third of our patients highlights the role of MCH-II proteins in immune tolerance [9, 46]. MHC-II deficiency also causes a complex disruption of mucosal immunity, presenting with inflammatory intestinal manifestations [47]. Thus, MHC-II deficiency should be considered a differential diagnosis in early-onset inflammatory bowel diseases. Neurologic problems were documented in one-third of our patients, consistent with previous studies that reported neurologic involvement, mainly in patients with RFXANK mutation [15, 22, 48]. Some of these findings could be attributed to viral infections, but the exact mechanisms of the neurologic phenotypes are unclear.
In this study, most patients displayed a reduced absolute CD4 + T cell count and absent HLA-DR expression on B lymphocytes and monocytes. The CD4 + T cell counts were normal in 27.7% of the patients, while they were functionally defective in all tested patients. Despite normal CD4 + T cells, these patients had clinical courses like others. In our study, 33.3% of patients had low B cell numbers. In other cohorts, 21 to 24% of the patients had fewer circulating B cells than expected [15, 49]. The reasons for this variability in immunologic phenotype in this setting are unknown. Genetic and environmental factors other than the level of HLA-DR expression level may explain the variable clinical expression [13, 15]. We could not exclude the presence of revertant mosaicism as the underlying cause.
We documented RFXANK, RFXAP, and RFX5 mutations in Iranian patients with MHC-II deficiency. Despite such genetic heterogeneity, these patients had a similar clinical presentation. About 72% of the patients had a RFXANK mutation, consistent with international observations. Six patients had the same homozygous c.162delG frameshift deletion in the RFXANK gene limited to the Iranian population. A founder effect was demonstrated about 1296 years ago. Also, we demonstrated the novel splice-site mutation (c.438 + 5G > A) in two patients with a milder form of the disease. Late onset of symptoms and milder course have been reported previously with other splicing RFXANK mutations [12].
HSCT is the only curative treatment for this IEI. However, HSCT had a limited success rate in these patients, not exceeding 50% [23, 41, 50,51,52,53]. Residual host immunity in MHC-II deficiency is sufficient to cause rejection [23, 50, 54, 55]. Alternatively, donor antigen-presenting cells could present donor antigens to recipient T cells leading to graft rejection.
Recent studies have shown an improved survival of up to 94% [49]. Multiple factors have contributed to this improvement, including the age at HSCT of less than 2 years, a meticulous graft selection strategy, improved graft manipulation methods, better supportive care, vigilant infection surveillance, and more effective antimicrobial therapies [49, 53, 54].
As with other studies, MHC-II-deficient patients undergoing HSCT seem to have persistently lower numbers of CD4 + T cells with a moderate decrease in naive CD4 + T cells. This finding is consistent with impaired thymic maturation due to defective MHC-II expression on thymic epithelia [9]. Despite post-HSCT CD4 + T cell lymphopenia, these patients display a normalization of antigen-specific T cell stimulation and antibody production in response to immunization antigens [15, 49].
In conclusion, MHC-II deficiency is not rare in Iran; it should be considered in the differential diagnosis of CID at any age. With the limited access to HSCT and its variable results in MHC-II deficiency, implementing genetic counseling and family planning for the affected families are mandatory. We have identified and dated a founder event responsible for the RFXANK c.162delG frameshift deletion limited to the Iranian population.
Data Availability
The NGS data have been submitted to GenBank; the accession numbers are pending.
Abbreviations
- ARD:
-
Ankyrin repeat domain
- APCs:
-
Antigen-presenting cells
- ANCA:
-
Antineutrophil cytoplasmic antibodies
- AIHA:
-
Autoimmune hemolytic anemia
- BCG:
-
Bacillus Calmette–Guerin
- BAL:
-
Bronchoalveolar lavage
- BMSC:
-
Bone marrow stem cells
- CSF:
-
Cerebrospinal fluid
- CADD:
-
Combined annotation-dependent depletion
- CID:
-
Combined immunodeficiency
- CT:
-
Computed tomography
- CMV:
-
Cytomegalovirus
- DTH:
-
Delayed-type hypersensitivity
- DBD:
-
DNA-binding domain
- ENT:
-
Ear, nose, and throat
- FTT:
-
Failure to thrive
- HSCT:
-
Hematopoietic stem cell transplantation
- HSV:
-
Herpes simplex virus
- iVDPV:
-
Immunodeficiency-associated vaccine–derived poliovirus
- IEI:
-
Inborn errors of immunity
- JIA:
-
Juvenile idiopathic arthritis
- MHC-II:
-
Major histocompatibility complex class II
- MRI:
-
Magnetic resonance imaging
- MRD:
-
Matched-related donor
- MAF:
-
Minor allele frequency
- MMRD:
-
Mismatched-related donor
- MRCA:
-
Most recent common ancestor
- MSC:
-
Mutation significance cutoff
- NDD:
-
Neurodevelopmental delay
- NLH:
-
Nodular lymphoid hyperplasia
- NTD:
-
N-terminal domain
- NLS:
-
Nuclear localization signal
- OPV:
-
Oral poliovirus vaccine
- PBSC:
-
Peripheral blood stem cells
- PEST:
-
Activation domain rich in acidic amino acids (P, E, S, and T)
- PHA:
-
Phytohemagglutinin
- SCID:
-
Severe combined immunodeficiency
- TCR:
-
T cell receptor
- VAPP:
-
Vaccine-associated paralytic poliomyelitis
- VDPV2:
-
Vaccine-derived poliovirus type 2
References
Viret C, Janeway C Jr. MHC and T cell development. Rev Immunogenet. 1999;1(1):91–104.
Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12(1):259–91.
Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331.
Villard J, Masternak K, Lisowska-Grospierre B, Fischer A, Reith W. MHC class II deficiency: a disease of gene regulation. Medicine. 2001;80(6):405–18.
Griscelli C, Lisowska-Grospierre B, Le Deist F, Durandy A, Marcadet A, Fischer A, et al. Combined immunodeficiency with abnormal expression of MHC class II genes. Clin Immunol Immunopathol. 1989;50(1 Pt 2):S140–8.
Griscelli C, Lisowska-Grospierre B, Mach B. Combined immunodeficiency with defective expression in MHC class II genes. Immunodefic Rev. 1989;1(2):135–53.
Reith W, Satola S, Sanchez CH, Amaldi I, Lisowska-Grospierre B, Griscelli C, et al. Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein. RF-X Cell. 1988;53(6):897–906.
Lisowska-Grospierre B, Durandy A, Virelizier JL, Fischer A, Griscelli C. Combined immunodeficiency with defective expression of HLA: modulation of an abnormal HLA synthesis and functional studies. Birth Defects Orig Artic Ser. 1983;19(3):87–91.
Ferrua F, Bortolomai I, Fontana E, Di Silvestre D, Rigoni R, Marcovecchio GE, et al. Thymic epithelial cell alterations and defective thymopoiesis lead to central and peripheral tolerance perturbation in MHCII deficiency. Front Immunol. 2021;12:669943.
Waldburger J-M, Masternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Landmann S, et al. Lessons from the bare lymphocyte syndrome: molecular mechanisms regulating MHC class II expression. Immunol Rev. 2000;178:148–65.
Masternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Reith W. Molecular genetics of the bare lymphocyte syndrome. Rev Immunogenet. 2000;2(2):267–82.
Prod’homme T, Dekel B, Barbieri G, Lisowska-Grospierre B, Katz R, Charron D, et al. Splicing defect in RFXANK results in a moderate combined immunodeficiency and long-duration clinical course. Immunogenetics. 2003;55(8):530–9.
Wiszniewski W, Fondaneche M-C, Le Deist F, Kanariou M, Selz F, Brousse N, et al. Mutation in the class II trans-activator leading to a mild immunodeficiency. J Immunol. 2001;167(3):1787–94.
Hanna S, Etzioni A. MHC class I and II deficiencies. J Allergy Clin Immunol. 2014;134(2):269–75.
Ouederni M, Vincent QB, Frange P, Touzot F, Scerra S, Bejaoui M, et al. Major histocompatibility complex class II expression deficiency caused by a RFXANK founder mutation: a survey of 35 patients. Blood J Am Soc Hematol. 2011;118(19):5108–18.
Bejaoui M, Barbouche M, Mellouli F, Largueche B, Dellagi K. Primary immunologic deficiency by deficiency of HLA class II antigens: nine new Tunisian cases. Arch Pediatr: Organe Officiel de la Soc Fr Pediatr. 1998;5(10):1089–93.
Ben-Mustapha I, Ben-Farhat K, Guirat-Dhouib N, Dhemaied E, Larguèche B, Ben-Ali M, et al. Clinical, immunological and genetic findings of a large Tunisian series of major histocompatibility complex class II deficiency patients. J Clin Immunol. 2013;33(4):865–70.
Djidjik R, Messaoudani N, Tahiat A, Meddour Y, Chaib S, Atek A, et al. Clinical, immunological and genetic features in eleven Algerian patients with major histocompatibility complex class II expression deficiency. Allergy Asthma Clin Immunol. 2012;8(1):1–5.
El Hawary RE, Mauracher AA, Meshaal SS, Eldash A, AbdElaziz DS, Alkady R, et al. MHC-II deficiency among Egyptians: novel mutations and unique phenotypes. J Allergy Clin Immunol: In Pract. 2019;7(3):856–63.
Wiszniewski W, Fondaneche M-C, Lambert N, Masternak K, Picard C, Notarangelo L, et al. Founder effect for a 26-bp deletion in the RFXANK gene in North African major histocompatibility complex class II-deficient patients belonging to complementation group B. Immunogenetics. 2000;51(4):261–7.
Al-Herz W, Zainal ME, Salama M, Al-Ateeqi W, Husain K, Abdul-Rasoul M, et al. Primary immunodeficiency disorders: survey of pediatricians in Kuwait. J Clin Immunol. 2008;28(4):379–83.
Klein C, Lisowska-Grospierre B, LeDeist F, Fischer A, Griscelli C. Major histocompatibility complex class II deficiency: clinical manifestations, immunologic features, and outcome. J Pediatr. 1993;123(6):921–8.
Saleem M, Arkwright P, Davies E, Cant A, Veys P. Clinical course of patients with major histocompatibility complex class II deficiency. Arch Dis Child. 2000;83(4):356–9.
Wiszniewski W, Fondaneche M-C, Louise-Plence P, Prochnicka-Chalufour A, Selz F, Picard C, et al. Novel mutations in the RFXANK gene: RFX complex containing in-vitro-generated RFXANK mutant binds the promoter without transactivating MHC II. Immunogenetics. 2003;54(11):747–55.
Cai YQ, Zhang H, Wang XZ, Xu C, Chao YQ, Shu Y, et al. A novel RFXANK mutation in a chinese child with MHC II deficiency: case report and literature review. Open Forum Infect Dis. 2020;7(8):ofaa314.
Abolnezhadian F, Dehghani R, Dehnavi S, Khodadadi A, Shohan M. A novel mutation in RFXANK gene and low B cell count in a patient with MHC class II deficiency: a case report. Immunol Res. 2020;68(4):225–31.
Abolnezhadian F, Saeedi-Boroujeni A, Iranparast S. MHC class II deficiency with normal CD4+ T cell counts: a case report. Iran J Allergy Asthma Immunol. 2018;17(6):594–600.
Farrokhi S, Shabani M, Aryan Z, Zoghi S, Krolo A, Boztug K, et al. MHC class II deficiency: report of a novel mutation and special review. Allergol Immunopathol (Madr). 2018;46(3):263–75.
Parvaneh N, Shahmahmoudi S, Tabatabai H, Zahraei M, Mousavi T, Esteghamati AR, et al. Vaccine-associated paralytic poliomyelitis in a patient with MHC class II deficiency. J Clin Virol. 2007;39(2):145–8.
Sheikhbahaei S, Sherkat R, Roos D, Yaran M, Najafi S, Emami A. Gene mutations responsible for primary immunodeficiency disorders: a report from the first primary immunodeficiency biobank in Iran. Allergy Asthma Clin Immunol. 2016;12:62.
Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med. 2019;216(9):2057–70.
Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–10.
Krawczyk M, Masternak K, Zufferey M, Barras E, Reith W. New functions of the major histocompatibility complex class II-specific transcription factor RFXANK revealed by a high-resolution mutagenesis study. Mol Cell Biol. 2005;25(19):8607–18.
Nekrep N, Geyer M, Jabrane-Ferrat N, Peterlin BM. Analysis of ankyrin repeats reveals how a single point mutation in RFXANK results in bare lymphocyte syndrome. Mol Cell Biol. 2001;21(16):5566–76.
Genin E, Tullio-Pelet A, Begeot F, Lyonnet S, Abel L. Estimating the age of rare disease mutations: the example of triple-A syndrome. J Med Genet. 2004;41(6):445–9.
Wang RJ, Al-Saffar SI, Rogers J, Hahn MW. Human generation times across the past 250,000 years. Sci Adv. 2023;9(1):eabm7047.
Laird KM, Briggs LL, Boss JM, Summers MF, Garvie CW. Solution structure of the heterotrimeric complex between the interaction domains of RFX5 and RFXAP from the RFX gene regulatory complex. J Mol Biol. 2010;403(1):40–51.
Villard J, Peretti M, Masternak K, Barras E, Caretti G, Mantovani R, et al. A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol Cell Biol. 2000;20(10):3364–76.
Chakraborty M, Sengupta A, Bhattacharya D, Banerjee S, Chakrabarti A. DNA binding domain of RFX5: interactions with X-box DNA and RFXANK. Biochim Biophys Acta (BBA)-Protein Proteomics. 2010;1804(10):2016–24.
Naamane H, El Maataoui O, Ailal F, Barakat A, Bennani S, Najib J, et al. The 752delG26 mutation in the RFXANK gene associated with major histocompatibility complex class II deficiency: evidence for a founder effect in the Moroccan population. Eur J Pediatr. 2010;169(9):1069–74.
Klein C, Cavazzana-Calvo M, Le Deist F, Jabado N, Benkerrou M, Blanche S, et al. Bone marrow transplantation in major histocompatibility complex class II deficiency: a single-center study of 19 patients. Blood. 1995;85(2):580–7.
Shaghaghi M, Shahmahmoodi S, Nili A, Abolhassani H, Madani SP, Nejati A, et al. Vaccine-derived poliovirus infection among patients with primary immunodeficiency and effect of patient screening on disease outcomes. Iran Emerg Infect Dis. 2019;25(11):2005–12.
Shahmahmoodi S, Mamishi S, Aghamohammadi A, Aghazadeh N, Tabatabaie H, Gooya MM, et al. Vaccine-associated paralytic poliomyelitis in immunodeficient children, Iran, 1995–2008. Emerg Infect Dis. 2010;16(7):1133–6.
El-Sayed ZA, El-Ghoneimy DH, Ortega-Martell JA, Radwan N, Aldave JC, Al-Herz W, et al. Allergic manifestations of inborn errors of immunity and their impact on the diagnosis: a worldwide study. World Allergy Organ J. 2022;15(6):100657.
Fazlollahi MR, Najmi M, Fallahnezhad M, Sabetkish N, Kazemnejad A, Bidad K, et al. Paediatric asthma prevalence: the first national population-based survey in Iran. Clin Respir J. 2019;13(1):14–22.
Hervé M, Isnardi I, Ng Y-s, Bussel JB, Ochs HD, Cunningham-Rundles C, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med. 2007;204(7):1583–93.
Posovszky C, Sirin M, Jacobsen E, Lorenz M, Schwarz K, Schmidt-Choudhury A, et al. Persisting enteropathy and disturbed adaptive mucosal immunity due to MHC class II deficiency. Clin Immunol. 2019;203:125–33.
Alharby E, Obaid M, Elamin MAO, Almuntashri M, Bakhsh I, Samman M, et al. Progressive ataxia and neurologic regression in RFXANK-associated bare lymphocyte syndrome. Neurol Genet. 2021;7(3):e586.
Lum SH, Anderson C, McNaughton P, Engelhardt KR, MacKenzie B, Watson H, et al. Improved transplant survival and long-term disease outcome in children with MHC class II deficiency. Blood. 2020;135(12):954–73.
Renella R, Picard C, Neven B, Ouachée-Chardin M, Casanova JL, Deist FL, et al. Human leucocyte antigen-identical haematopoietic stem cell transplantation in major histocompatiblity complex class II immunodeficiency: reduced survival correlates with an increased incidence of acute graft-versus-host disease and pre-existing viral infections. Br J Haematol. 2006;134(5):510–6.
Siepermann M, Gudowius S, Beltz K, Strier U, Feyen O, Troeger A, et al. MHC class II deficiency cured by unrelated mismatched umbilical cord blood transplantation: case report and review of 68 cases in the literature. Pediatr Transplant. 2011;15(4):E80–6.
Antoine C, Müller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet. 2003;361(9357):553–60.
Bonduel M, Staciuk R, Figueroa C, Oleastro M, Gamba C, Rossi J, et al. Unrelated cord blood transplantation and reduced-intensity conditioning regimen for graft failure in a child with major histocompatibility complex class II deficiency. Bone Marrow Transplant. 2009;43(10):817–8.
Al-Mousa H, Al-Shammari Z, Al-Ghonaium A, Al-Dhekri H, Al-Muhsen S, Al-Saud B, et al. Allogeneic stem cell transplantation using myeloablative and reduced-intensity conditioning in patients with major histocompatibility complex class II deficiency. Biol Blood Marrow Transplant. 2010;16(6):818–23.
Small TN, Qasim W, Friedrich W, Chiesa R, Bleesing JJ, Scurlock A, et al. Alternative donor SCT for the treatment of MHC class II deficiency. Bone Marrow Transplant. 2013;48(2):226–32.
Author information
Authors and Affiliations
Contributions
Conceptualization: Nima Parvaneh. Methodology: Nima Parvaneh, Mohammad Shahrooei, Laurent Abel. Formal analysis and investigation: Mohadese Sadat Mousavi Khorshidi, Yoann Seeleuthner, Zahra Chavoshzadeh, Maryam Behfar, Amir Ali Hamidieh, Hosein Alimadadi, Roya Sherkat, Tooba Momen, Nasrin Behniafard, Shabnam Eskandarzadeh, Mahboubeh Mansouri, Mahdiyeh Behnam, Mohadese Mahdavi, Maryam Heydarazad Zadeh, Mehdi Shokri, Fatemeh Alizadeh, Mahshid Movahedi, Mana Momenilandi, Mohammad Keramatipour, Mohammad Shahrooei, Jean-Laurent Casanova, Aurélie Cobat, Laurent Abel, Nima Parvaneh. Writing — original draft preparation: Mohadese Sadat Mousavi Khorshidi, Yoann Seeleuthner, Nima Parvaneh. Writing — review and editing: Nima Parvaneh, Jean-Laurent Casanova, Laurent Abel, Aurélie Cobat. Supervision: Nima Parvaneh, Mohammad Shahrooei, Jean-Laurent Casanova, Laurent Abel, Aurélie Cobat.
Corresponding author
Ethics declarations
Ethics Approval
The Children’s Medical Center Institutional Ethics Committee, affiliated with the Tehran University of Medical Sciences, approved the study.
Consent to Participate/Consent for Publication
Informed consent for participating in the study and publication of data was obtained from the patients or their parents.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mousavi Khorshidi, M.S., Seeleuthner, Y., Chavoshzadeh, Z. et al. Clinical, Immunological, and Genetic Findings in Iranian Patients with MHC-II Deficiency: Confirmation of c.162delG RFXANK Founder Mutation in the Iranian Population. J Clin Immunol 43, 1941–1952 (2023). https://doi.org/10.1007/s10875-023-01562-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-023-01562-z