
Abstract
Immune responses to coronavirus disease 2019 (COVID-19) mRNA vaccines in primary antibody deficiencies (PADs) are largely unknown. We investigated antibody and CD4+ T-cell responses specific for SARS-CoV-2 spike protein (S) before and after vaccination and associations between vaccine response and patients’ clinical and immunological characteristics in PADs. The PAD cohort consisted of common variable immune deficiency (CVID) and other PADs, not meeting the criteria for CVID diagnosis (oPADs). Anti-S IgG, IgA, and IgG subclasses 1 and 3 increased after vaccination and correlated with neutralization activity in HCs and patients with oPADs. However, 42% of CVID patients developed such responses after the 2nd dose. A similar pattern was also observed with S-specific CD4+ T-cells as determined by OX40 and 4-1BB expression. Patients with poor anti-S IgG response had significantly lower levels of baseline IgG, IgA, CD19+ B-cells, switched memory B-cells, naïve CD8+ T-cells, and a higher frequency of EM CD8+ T-cells and autoimmunity compared to patients with adequate anti-S IgG responses. Patients with oPADs can develop humoral and cellular immune responses to vaccines similar to HCs. However, a subset of CVID patients exhibit impairment in developing such responses, which can be predicted by the baseline immune profile and history of autoimmunity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary antibody deficiencies (PADs) are a diverse group of disorders that account for 50–60% of all primary immunodeficiencies [1,2,3]. PADs include common variable immune deficiency (CVID), IgG subclass 2 deficiency (IgG2D), IgG deficiency (IgGD), and specific antibody deficiency (sAbD). It has been shown that many PAD patients are unable to mount an effective antibody response to pathogens and/or vaccines [3, 4]. Patients with immunodeficiencies have an increased risk for poor clinical outcomes with coronavirus disease 2019 (COVID-19) [5, 6]. Thus, the inability of many of these patients to mount a substantial immune response to vaccination poses a particular challenge as vaccines are currently the most effective protection against COVID-19.
COVID-19 vaccines induce both humoral and cellular immunity as measured by SARS-CoV-2 spike protein (S)-specific IgG antibodies and responsive CD4+ T-cells. Patients with various primary immunodeficiency disorders including inborn errors of immunity (IEI) and CVID were found to have reduced levels of SARS-CoV-2 S-specific IgG responses [7,8,9,10,11]. For instance, anti-S antibodies were detected in only 27 of 46 patients with IEI (58.7%) after 1 dose of COVID-19 mRNA vaccine and in 63 of 74 fully immunized patients with IEI (85.1%). The impaired development of SARS-CoV-2 S-specific IgG response was associated with previous use of rituximab therapy, reduced baseline B and T cells counts, and specific gene defects including X-linked agammaglobulinemia (XLA), autoimmune lymphoproliferative syndrome (ALPS)-like disease, and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) [7,8,9,10,11]. In addition to humoral immunity, patients with ALPS-like disease and NF-κB1 mutations failed to mount T cell immune responses to COVID-19 vaccines [7, 10]. In patients with CVID, the development of both humoral and cellular immune responses to COVID-19 vaccines was delayed and reduced when compared to healthy controls (HC) [8].
Although studies indicate impaired vaccine responses in patients with PADs including CVID and specific gene defects, the immunogenicity of COVID-19 vaccines in patients with humoral immunodeficiencies such as IgGD, IgG2D, and sAbD remains largely unknown. Patients with such immunodeficiencies are known to have significantly increased sinopulmonary infections and non-infectious clinical manifestations including autoimmunity and malignancy [12, 13]. To address this critical point, we evaluated the humoral and CD4+ T-cell immune responses to COVID-19 mRNA vaccines by analyzing anti-S IgG, IgA, IgM, and IgG subclasses (1, 2, 3, and 4), plasma neutralizing activity against the SARS-CoV-2 USA-WA 1/2020 in vitro, and S-specific CD4+ T-cell responses in patients with a wide spectrum of PADs prior to and following the 1st and 2nd doses of COVID−19 mRNA vaccines. We correlated these findings with baseline immunoglobulin levels, immune cell profiles, and comorbidities to identify the possible predictors for poor COVID-19 vaccine responses in these patients.
Methods
Human Subjects
The study was reviewed and approved by the Yale University Institutional Review Board (IRB). The Informed consent for the study was obtained from all participating study subjects.
Patients with PADs (n = 22) were recruited from Yale Immunodeficiency clinics. We categorized patients into one of two groups, a group of CVID or a combined group of IgGD, IgG2D, and sAbD referred to as other PAD (oPAD). HCs (n = 15) were recruited as previously described [14]. After obtaining informed consent, samples were collected at three time points: prior to vaccination, three (Pfizer-BioNTech) or four (Moderna) weeks after the 1st and 2nd doses of the Pfizer-BioNTech or Moderna COVID-19 mRNA vaccine. Clinical history and laboratory data were collected using the EPIC electronic medical record (Epic Systems Corporation, Verona, WI, USA).
Peripheral Mononuclear Cell Purification
Peripheral blood was collected into sodium heparin tubes. Plasma was separated by centrifuging the tubes at 1800 rpm for 10 min and stored in 1.8 mL cryotubes (Thermo Fisher Scientific, Waltham, MA, USA) at − 80 °C until further analysis. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque ™ PREMIUM sterile solution (Cytiva, Marlborough, MA, USA). After isolation, the cells were cryopreserved in fetal bovine serum Gibco ™ (FBS, Thermo Fisher Scientific) with 10% dimethyl sulfoxide ACS (DMSO; MP Biomedicals, LLC, Solon, OH, USA) and stored in vapor phase liquid nitrogen prior to use.
Enzyme-Linked Immunosorbent Assay (ELISA)
The humoral response to the vaccines was evaluated by analyzing anti–SARS-CoV-2 spike protein receptor-binding domain (SRBD) antibodies (IgG, IgA, IgM, and IgG subclasses 1, 2, 3, and 4) and anti-SARS-CoV-2 nucleocapsid IgG in plasma. High-binding 96-well clear flat bottom polystyrene plates (Costar®, Corning, Durham, NC, USA) were coated with 1 µg/ml of SARS-CoV-2 Spike S1 + S2 ECD-His Recombinant Protein (Sino Biological, Chesterbrook, PA, USA) or SARS-CoV-2 Nucleocapsid-His recombinant protein (Sino Biological, Chesterbrook, PA, USA) in 0.05 M carbonate solution (sodium bicarbonate buffer capsule; Millipore Sigma, Burlington, MA, USA) by incubating overnight at 4 °C. Following the incubation, coating solution was discarded, and plates were blocked for 1 h with 3% (w/v) non-fat dry milk Omniblok ™ (AmericanBio, Canton, MA, USA) in PBS 1X at room temperature. The plates were then washed two times with ELISA wash buffer containing 0.05% (v/v) Tween 20 in PBS 1X. Plasma samples were diluted in 1% (w/v) Omniblok ™ in wash buffer (1:200 for IgG, IgA, IgM, IgG1; 1:10 for IgG2, IgG3, and IgG4), pipetted into appropriate wells in duplicates, and incubated for 1 h at room temperature. Positive control (plasma know to have + anti-S IgG) and a blank well were included in each plate. The plates were then washed 5 times with the ELISA wash buffer, added with secondary antibodies diluted at 1:2000 (IgG) and 1:1000 (IgA, IgM, IgG1, IgG2, IgG3, IgG4) at 75µL per well and incubated for 1 h at room temperature. Plates were washed 6 times with the ELISA wash buffer and developed with TMB Substrate solution (BD OptEIA, BD Biosciences, San Diego, CA, USA) by incubating for 2–4 (IgG), 8–12 (IgA, IgM and IgG1), and 15–20 min (IgG2, IgG3, IgG4) in the dark. After adding stop solution (2 N sulfuric acid, R&D Systems, Minneapolis, MN, USA), the optical density (OD) of individual samples was measured at 450 nm using the Synergy ™ HT multi-detection microplate reader (Bio-Tek Instruments, INC., Winooski, VT, USA). The results of duplicate samples were averaged. The OD value from the blank was subtracted from the average sample ODs to obtain the absolute absorbance reading. Arbitrary Unit (AU) was determined by dividing absolute absorbance reading of subjects by the absorbance reading of positive control.
Flow Cytometry
PBMCs were thawed and placed in 15-mL Eppendorf tubes with 9 mL of RPMI Medium 1640 (Gibco™) supplemented with 1% penicillin, streptomycin, and glutamate and 10% fetal bovine serum (RPMI-CM) and centrifuged at 1800 rpm for 10 min. Following centrifugation, cells were resuspended in 2 mL of RPMI-CM in 5 mL round-bottom polypropylene tubes (Falcon®, Corning) and incubated at 37 °C for 7–8 h. Cells were counted and pipetted at 1 × 106 cells per well for T-cell stimulation. For the latter, cells were incubated for 18–20 h at 37 °C in 96-well clear round bottom cell culture microplate (Cat# 353077, Falcon®) with or without 2 µg/mL anti-CD28/CD49d antibody (BD Fastimmune, BD Biosciences, Cat# 347690), SARS-CoV-2 Prot_S (Miltenyi Biotec, Cat#130–126-700, Bergisch Gladbach, Germany), or SARS-CoV-2 Prot_N (Miltenyi Biotec, Cat#130–126-698) in TexMACS™ media (Miltenyi Biotec, Cat#130–097-196). After incubation, cells were harvested and incubated for 10 min with LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (Thermo Fisher Scientific, Cat# L10119) at room temperature to determine viable cells. Then the viable cells were analyzed for CD3, CD4, CCR7, CD45RA, OX40, and 4-1BB. In addition, unstimulated viable cells were stained with appropriate antibodies for T- and B-cell subset analysis. The following fluorochrome-labeled antibodies were used: BUV 395 Anti-hCD3 (BD Horizon, BD Biosciences, Clone SK7, Cat#564001, 1:150), BV 711 anti-hCD4 (BioLegend, San Diego, CA, USA, Clone SK3, Cat #344648, 1:150), BV510 anti-hCD8 (BioLegend, Clone SK1, Cat#344732, 1:150), PE-Cy5 anti-hCD45RA (BD Pharmingen, Cat#555490, 1:100), PE-Cy7 anti-hCCR7 (BD Biosciences, Cat#557648, 1:100), FITC anti-hCD134 OX40 (BioLegend, Clone Ber-ACT35, Cat#350006, 1:50), PE anti-hCD137 4-1BB (BioLegend, Clone 4B4-1 Cat#309804, 1:50), anti-hCD185 CXCR5 (Invitrogen, Waltham, MA, USA, Clone MU5UBEE Cat#14–9185-82, 1:50), Alexa Fluor 488 anti-hCD279 (BioLegend, Clone EH12.2H7, Cat#329935, 1:50), Alexa Fluor 700 anti-hCD19 (BioLegend, Clone H1B19 Cat#302225, 1:100), anti-hCD27 PB (Invitrogen, Clone 0323 Cat#48–0279-41, 1:50), PE/Cy7 anti-hCD21 (BioLegend, Clone BU32 Cat#354911, 1:50), PE anti-hIgM (BioLegend, Clone MHM-88, Cat#314507, 1:50), and Alexa Fluor 488 anti-hIgD (BioLegend, Clone IA6-2, Cat#348215, 1:50). Flow cytometry analysis was done using an LSR II instrument with FACSDiva (BD Biosciences). Flow cytometry data was analyzed using FlowJo v10 software (BD, Ashland, OR, USA). Spike RBD-specific CD4 T-cell activation was determined by measuring effector memory CD4+ T cells expressing OX40 and 4-1BB.
SARS-CoV-2 Surrogate Viral Neutralization Assay
The SARS-CoV-2 isolate USA-WA1-/2020 was used to study neutralization activity of patient plasma. The strain was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281, kindly provided by Barney Graham at Vaccine Research Center of NIH. This was then cultured in Vero-hACE2-TMPRSS2 which were prepared at a concentration of 1.25 cells/mL in 10 mL Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific, Cat#11054020) with 10% heat-inactivated fetal bovine serum (FBS) and 1% GlutaMAX (Thermo Fisher Scientific, Cat# 35050061). Subject plasma samples from three different time points were inactivated by incubation at 56 °C for 1 h. Samples were diluted three-fold in 8 serial dilutions in a 384-well plate (10 to 21,870 dilution; Corning, Cat#3764). Ten microliter of diluted plasma was incubated with an equal volume of USA-WA1/2020 at a multiplicity of infection (MOI) of 0.5 at 37 °C for 30 min. Ten microliter of Vero-hACE2-TMPRSS2 cells were transferred to appropriate well with virus/plasma mix, and the mixture was incubated at 37 °C for another 72 h to allow infection to proceed. 12.5 μL of Cell Titer Glo™ (Promega, Madison, WI, USA, Cat#G9241) was pipetted to each well, and plate was incubated at room temperature for 10 min before luminescence was read using Cytation5. Concentration to inhibit 50% of infection (IC50) was calculated using non-linear regression compared to mock-infected samples.
Statistical Analysis
Descriptive data were presented as means ± standard deviations (SD)s. Continuous variables were analyzed using the Student t test or one-way ANOVA with the Dunnett’s test for multiple comparisons as appropriate. Categorical variables were analyzed using the Chi-square test. Data were analyzed with SPSS version 28.0 (IBM, Armonk, NY, USA) and Prism 9 (GraphPad Software, Inc., San Diego, CA, USA). P values of 0.05 or less were considered statistically significant.
Results
Cohorts and Patient Characteristics
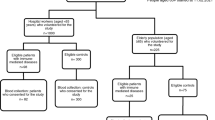
Fifteen HCs and 22 patients with PADs were recruited from the pediatric or adult immunodeficiency clinics at Yale. One HC and six patients with PADs had SARS-CoV-2 infection confirmed with positive nucleic-acid assay prior to vaccination (Table 1). The rest of HCs and patients without positive nucleic acid assay for SARS-CoV-2 did not present with increased IgG levels for SARS-CoV-2 nucleocapsid (Fig. S2B). We collected samples from 10 HCs and 17 patients with PAD at baseline and following each COVID-19 mRNA vaccination (Fig. 1). For the remaining subjects, we had samples following the 1st and 2nd doses (n = 3 HCs and 2 patients), before vaccination and after the 2nd dose (n = 1 HC and 1 patients) or after the 2nd dose (n = 1 HC and 2 patients). The patients with PADs met the diagnostic criteria for CVID (n = 12), IgG deficiency (IgGD, n = 5), IgG subclass 2 deficiency (IgG2D, n = 4), and specific antibody deficiency (sAbD, n = 1) [13]. The demographic and clinical characteristics of patients are described in Table 1. Of note, patient 2 had confirmed PLC-γ-2 mutation (Table 1). Rest of the patients did not have confirmed diagnosis of monogenetic mutation. Patient 9 received rituximab about 6 years ago, and the patient showed recovered B cell count (CD19+ cells) of 149 cell/ul. The mean ages of the studied groups were similar (HCs, CVID, and oPAD, mean ± SD; 40.5 ± 11.1, 42.7 ± 21.5, and 55.0 ± 18.3, respectively). The gender ratios were similar in HCs and CVID patients (percent male: 47% and 39%, respectively); however, the proportion of males in patients with oPAD was lower than in HCs and CVID patients (percent male: 20%, 47%, and 39%, respectively, p = 0.0009).

Study design and schedule for sample collection. Peripheral blood was collected from healthy controls, patients with CVID,and other PADs at three time points: before, 3 (Pfizer-BioNTech) or 4 (Moderna) weeks after the 1st and 2nd doses of the Pfizer−BioNTech or Moderna COVID-19 mRNA vaccine
Patients with CVID Had a Slower Rise and Achieved a Lower Peak in Anti-S IgG, IgA, IgG Subclasses 1 and 3 Titers Compared to HCs and oPAD Patients After COVID-19 mRNA Vaccination
Samples collected pre-vaccination, 3 weeks or 4 weeks after the 1st and 2nd doses of Pfizer-BioNTech or Moderna vaccine, were applied to anti-S ELISA as described in the “Methods” section. After the first dose of the vaccine, all HCs and oPADs patients developed anti-S IgG, which continued to increase after the second dose (Fig. 2). The mean and two SDs of anti-S IgG titers in HCs were 0.5 ± 0.1, respectively, after the 2nd dose of vaccination. Based on these values, patients who had IgG titers equal or less than 0.3 were classified as poor vaccine responders, and 5 out of 12 CVID patients (42%) met this criterion. Of note, we measured antibody response of two CVID patients after the 3rd dose of vaccination. They received the 3rd dose about 4 months after the 2nd dose. These two CVID patients initially presented with poor antibody response after the 1st and 2nd doses and remained unresponsive after the 3rd dose (Fig. S1). Anti-S IgG subclass 1 had a similar pattern as anti-S IgG. Anti-S IgA and anti-S IgG3 also increased in most HCs and oPAD patients following vaccination (Fig. 2) although anti-S IgG2, IgG4, and IgM did not demonstrate this incline (Fig. 2). Patients with SARS-CoV-2 infection prior to vaccination (marked with red dots in Fig. 2) had higher antibody titers in pre-vaccination samples which increased after the 1st dose but either plateaued or decreased after the 2nd dose (Fig. 2). Next, we compared antibody titer increase between pre and after 1st and 2nd doses of vaccines only among the patients who were not previously infected with SARS-CoV-2. While both HCs and oPAD showed a statistically significant increase in anti-S IgG and IgG1 after the 1 and 2 doses, the CVID patient cohort showed statistically significant increase in anti-S IgG and neutralization activity only after the 2nd dose (Fig. 3). Neutralization activity was significantly increased after 1st and 2nd doses in HCs but only after 2nd dose in PAD patients (Fig. 3). In HCs, anti-S IgG3 demonstrated a significant increase following both doses. All three cohorts showed an increase in anti-S IgA titer after each dose though it did not meet statistical significance. Of note, the antibody responses were similar between HCs who received Pfizer and Moderna mRNA vaccines (data not shown).

Changes in SARS-CoV-2 serology in healthy controls (HCs), patients with CVID, and other PADs (oPADs) before and after the 1st and 2nd doses of COVID-19 mRNA vaccines. The changes of anti-S IgG, IgA, IgM, IgG subclasses 1, 2, 3, and 4 and neutralization activity to SARS-CoV-2 USA-WA 1/2020 before and after the 1st and 2nd doses of vaccines are shown in healthy controls (HCs), patients with CVID, and other PADs (oPAD). Subjects who had SARS-CoV-2 infection prior to vaccinations were marked as red

The development of humoral immune responses to COVID19 mRNA vaccines is distinct in healthy controls (HCs), patients with CVID, and other PADs (oPAD). Scatter graphs showing anti-S IgG, IgA, IgM, IgG subclasses 1, 2, 3, and 4, and neutralization activity to SARS-CoV-2 USA-WA 1/2020 before and after 1st and 2nd doses of vaccines in healthy controls (HCs), patients with CVID, or other PADs (oPADs) who previously did not have SARS-CoV-2 infection. P values were obtained by ANOVA followed by post hoc analysis with the Dunnett’s test
Neutralization Activity Against the SARS-CoV-2 WA 1/2020 In Vitro Correlated with Anti-S IgG, IgA and IgG Subclasses 1 and 3
Both HCs and study subjects displayed an increase in plasma neutralization activity after the 1st and/or 2nd dose of the vaccines (Figs. 2 and 3). We then determined the association between the fold change in neutralization activity and anti-S immunoglobulins measured at baseline and after the 2nd dose only in patients without prior SARS-CoV-2 infection. Fourteen patients without prior SARS-CoV-2 infection and with available data (neutralization activity and anti-S immunoglobulins levels of prior and after 2nd dose of vaccination) were included in this analysis. We found an association between fold change of neutralization activity and anti-S IgG in both HCs and PADs (anti-S IgG, HC: r = 0.7, p = 0.01, PAD: r = 0.6, p = 0.01) (Fig. 4). For the fold change of anti-S IgG1, only HC showed significant association with the fold change of neutralization although PADs had a trend of association (anti-S IgG1, HC: r = 0.8, p = 0.004, PAD: r = 0.5, p = 0.06) (Fig. 4). A similar association was also noted between fold change of neutralization activity and anti-S IgA titers in HC (IgA, HC: r = 0.8, p = 0.004) but not in PADs (Fig. 4). IgG subclasses 2 and 4 and IgM had no relationship with neutralization activity (Fig. 4).

Correlation of anti-S immunoglobulin titers with neutralization activity to SARS-CoV-2 USA-WA 1/2020 in healthy controls (HCs) and all PAD patients. Anti-S IgG titers are shown as fold changes between pre-vaccination and anti-S IgG after 2nd dose of vaccines. The correlation for fold changes of anti-S IgG, IgG subclasses (1, 2, 3, and 4), IgA, and IgM and neutralization activity are shown in HCs and all PAD patients. P values were obtained by Pearson correlation analysis
SARS-CoV-2 Spike Peptides Specific CD4+ T-Cell Response Showed Similar Trend as Humoral Response to COVID-19 mRNA Vaccines
We next studied CD4+ T-cell response specific for SARS-CoV-2 S or nucleocapsid peptides before and after COVID-19 mRNA vaccine. Previous studies identified CD4+ T cells specific for antigens including S protein in peripheral blood based on combinational expression of activation-induced markers (AIM) such as OX40 and 4-1BB [15,16,17]. Here, we took the same approach to identify CD4+ T cells to antigenic stimulation including S and nucleocapsid peptides. Thawed PBMC showed > 95% viability after in vitro stimulation with S or nucleocapsid peptides. Viable cells were gated for OX40 and 4-1BB expressing effector memory (EM) CD4+ T-cells (CD3+CD4+CCR7−CD45RA±) (Fig. 5A) which were specific for spike peptides or nucleocapsid specific EM CD4+ T cells after stimulation with respective antigen. Similar to humoral response, HCs and oPAD patients had an increase in the frequency of S-specific CD4+ T cells after the 1 dose of the vaccine although the increase in oPAD patients did not reach the level of significance. In CVID patients, S-specific CD4+ T-cells did not increase after the first dose of the vaccine. All three groups had an incremental increase in S-specific CD4+ T cells after each dose (Fig. 5B and C). We also evaluated CD4+ T-cell responses to SARS-CoV-2 nucleocapsid protein which is present in live virus but not in COVID-19 mRNA vaccine. Subjects with prior SARS-CoV-2 infection showed an increase in nucleocapsid-specific CD4+ T cells which did not change after vaccination (Fig. S2). Patients without known history of prior SARS-CoV-2 infection did not have an increase in nucleocapsid-specific CD4+ T cells (Fig. S2). In addition to the EM population, we analyzed S protein-specific cells in the central memory (CM) CD4+ T-cell population. The frequency of this cells was quite small ranging from 0.05 to 0.3% of the total CM population and did not change with vaccination (data not shown).

Flow cytometric analysis of S-specific OX40 and 4-1BB expressing CD4+ T cells in healthy controls (HC), patients with CVID, and other PADs before and after the 1st and 2nd doses of COVID-19 mRNA vaccines. A Representative gating strategy for S-specific OX40+ and 4-1BB+ CD4+ T cells. After excluding dead cells (7-AAD+ cells) from total lymphocytes, CCR7−CD45RA± effector memory (EM) cells were gated from the CD3+CD4+ T cell population followed by gating OX40 and 4-1BB expressing cells. B Trends and C mean ± SD for the frequency of OX40+ and 4-1BB+ cells in EM CD4+ T cells are shown pre and after the 1st (1) and 2nd (2) doses of vaccines (C) in healthy controls (HC), patients with CVID and other PADs (oPAD). P values were obtained by ANOVA followed by post hoc analysis with the Dunnett’s test
Lower Baseline IgG and IgA, a Lower Proportion of B Cells Including Switched Memory B cells, Higher Proportion of Memory CD8 T Cells, and an Autoimmune History Were Associated with Poor COVID-19 mRNA Vaccine Response
We measured CD8+ T-cell subsets (naïve, central memory (CM), effector memory (EM), and CD45RA+ EM), CD19+ B-cell subsets (CD27−CD21low cells and CD27+IgD−IgM− switched memory B cells (smB)), and circulating T-follicular helper (cTfh) cells (CD3+CD4+CD45RA− CCR7−CXCR5+PD-1+ and PD-1−) from unstimulated PBMCs (Fig. S3). The frequency of individual cell subsets was compared between vaccine responders vs. poor responders. The levels of baseline IgG (mean ± SD, 671 ± 240 and 228 ± 119 mg/dL, p < 0.0001), IgA (110 ± 78 and 47 ± 28 mg/dL, p = 0.001), IgG subclass 1 (mean ± SD, 443 ± 148 and 99 ± 86 mg/dL, p = 0.01), and IgG subclass 2 (mean ± SD, 163 ± 105 and 43 ± 22 mg/dL, p = 0.02) were significantly lower in poor responders than in responders (Table 2). The frequency of CD19 (mean (%) ± SD, 8 ± 5 and 5 ± 2, p = 0.02), smB cells (mean (%) ± SD, 23 ± 12 and 9 ± 2, p = 0.004), and naïve CD8 + T cells (mean (%) ± SD, 36 ± 21 and 27 ± 18, p = 0.02) was significantly lower in poor responders (Table 2), while the frequency of EM CD8+ T cells (mean (%) ± SD, 26% ± 12 and 30 ± 12%, p = 0.047) was significantly higher in poor responders. The poor responders had a trend towards the increased frequency of CD19+CD27−CD21low B (mean (%) ± SD, 38 ± 27, and 55 ± 26%, p = 0.166) and EMRA CD8+ T cells (mean (%) ± SD, 24 ± 20, and 32 ± 14%, p = 0.126), which have been shown to be associated with systemic autoimmunity and immunosenescence, respectively, compared to responders (Table 2) [18]. Poor responders had a higher frequency of autoimmune disorders (80% vs. 17%, p = 0.017) than responders.
Discussion
Although it has been shown that patients with PADs who meet criteria for CVID and/or carry specific gene defects showed reduced responses to COVID-19 mRNA vaccines, it remains largely unknown whether reduced immunogenicity is an important consideration for patients with other PADs such as IgGD, IgG2D, and sAbD. In this study, we analyzed anti-S IgG, IgA, IgM, and IgG subclasses (1, 2, 3, and 4), neutralization activity against SARS-CoV-2 USA-WA 1/2020 in vitro, and S-specific CD4+ T-cell responses in patients with a wide spectrum of PADs prior to vaccination and after the 1st and 2nd doses of the COVID-19 mRNA vaccines. We also looked at association between patients’ baseline immune profiles and comorbidities and efficacy of vaccine response. Our study shows that patients with IgGD, IgG2D, and sAbD develop anti-S IgG and IgG1 responses at the levels comparable to HCs following the 1st and 2nd doses of the COVID-19 mRNA vaccines. However, these responses were found in only some patients with CVID after the 2nd dose of the vaccines. A similar pattern was also observed in measuring neutralization activity against SARS-CoV-2 USA-WA 1/2020, and S protein-specific CD4+ T-cell responses. As defined based on the mean and two SDs of anti-S IgG titers in HC, the poor vaccine responders among PAD patients were those with autoimmune diseases and low levels of baseline IgG, IgG1, IgG2, CD19+ B, switched memory B cells, naïve CD8+ T cells, and higher levels of EM CD8+ T cells.
PADs are a diverse group of disorders including CVID, IgGD, IgG2D, and sAbD. Most patients with CVIDs are unable to produce an effective antibody response to pathogens or vaccination [3, 4]. In fact, the International Consensus Document 2016 criteria for CVID require impaired vaccine responses for diagnosis, unless the patient has profound hypogammaglobulinemia (IgG < 1 g/L)[19], although Ameratunga et al. suggested criteria not requiring impairment in vaccine responses to establish a diagnosis of CVID [20], recognizing that some patients with CVID can respond to vaccines. In patients who did not meet criteria for CVID in the New Zealand hypogammaglobulinemia study (NZHS), vaccine responses varied depending on the type of vaccine. Most patients enrolled in this study had excellent responses to Haemophilus influenzae type B (HIB) vaccine and tetanus toxoid but not to diphtheria toxoid or Streptococcus pneumoniae [21]. In our study, we noticed an increase in the levels of anti-S IgG and IgG1 in both HC and patients with oPAD including IgGD, IgG2D, and sAbD after the 1st and 2nd doses of the COVID-19 mRNA vaccines. However, some patients with CVID failed to mount significant humoral immune responses as measured by anti-S IgG and IgG1 levels after the 1st dose of the vaccines. With the 2nd dose, only 7 of 12 (58%) CVID patients achieved significant vaccine responses based on anti-S IgG response in comparison to HC. These findings are in line with the results of a recent study that analyzed immune responses to COVID-19 mRNA and DNA vaccines in patients with inborn errors of immunity, including disorders encompassing STAT3 dominant negative mutations (n = 26), APECED (n = 14), CVID (n = 7), and hypogammaglobulinemia (n = 5)[9]. Anti-S antibodies were detected in 27 of 46 patients (58.7%) after the 1st dose of mRNA vaccine and in 63 of 74 fully immunized patients (85.1%) [9]. Similar findings were also reported by other groups studying COVID-19 mRNA vaccine responses in patients with different types of PIDs [7,8,9,10,11]. In 18 CVID subjects receiving 2-dose anti-SARS-CoV-2 vaccines, 83% of the patients had S-specific antibodies after completing vaccination [8]. These findings indicate that a substantial proportion of patients with PADs, including CVID and inborn errors of immunity, cannot develop anti-S IgG responses following COVID-19 mRNA vaccination. Since these five CVID patients who did not present with anti-S antibody response after vaccinations were all receiving IgG replacement therapy, it is unlikely that passive immunization from IgG replacement therapy affected the results of anti-S antibody analysis in our patients. We also measured anti-S IgG response after the 3rd dose of mRNA vaccines in two CVID patients who initially presented with poor response to first two doses of COVID-19 mRNA vaccines. These patients continued to have poor response after the 3rd dose of mRNA vaccines, suggesting that CVID patients who have failed to mount antibody response to first two doses of mRNA vaccines less likely develop antibody response to subsequent dose(s) of COVID-19 mRNA vaccines.
We noticed increased levels of anti-S-specific IgG, IgG1, IgG3, and IgA in HC after COVID mRNA vaccination although patients with CVID and oPAD had increased levels of only anti-S-specific IgG and IgG1. The increased levels of anti-S-specific IgG likely reflect anti-S protein-specific IgG1 since IgG1 is the most abundant IgG subclass accounting for approximately 60% of total IgG [22]. Our observations are in line with the principle that COVID-19 mRNA vaccines induce the expression of SARS-CoV-2 spike protein, which triggers B-cells receiving T-cell help through MHC-class II, resulting in IgG1 and/or IgG3 class switching [22]. It may be beneficial to have class switching to several Ig classes and IgG subclasses given the different, albeit partially overlapping, functions of each. For instance, IgG1 and IgG3 potently activate complement by binding to C1q, while secretory IgA can provide mucosal immunity to microorganisms, including viruses. Furthermore, monomeric IgG3 binds more efficiently than monomeric IgG1 to FcγRIIa, FcγRIIIa and FcγRIIIb [23]. The relatively short half-life of IgG3 supports the potent pro-inflammatory function of IgG3 as it can limit the potential of excessive inflammatory responses. Of interest, patients with COVID-19 who required intensive care unit (ICU) admission had elevated levels of IgG3 compared to those treated on the floor or as outpatients [24].
In our study, we assessed titers of all immunoglobulin serotypes and IgG subclasses against SARS-CoV-2 spike protein as well as plasma neutralization activity against SARS-CoV-2 USA-WA 1/2020 strain before and after immunization with COVID-19 mRNA vaccines. A previous study showed that S-specific antibodies, primarily IgM and IgG1 in convalescent plasma of COVID-19 patients, exhibited neutralization activity to pseudovirus bearing the SARS-CoV-2 spike protein [25]. IgM and IgG1 were found to contribute most to SARS-CoV-2 neutralization [25]. Indeed, IgG1 and IgG3 subclasses are the main IgG subclasses induced by natural SARS-CoV-2 infection and COVID-19 vaccination [26, 27]. In our study, we found gradually rising titers of anti-S IgG and IgG1 in HC and patients with oPAD after the 1st and 2nd doses of the COVID-19 mRNA vaccines. Anti-S IgG3 also increased in HC but not in patients with CVID or oPAD. To the best of our knowledge, this is the first study to demonstrated neutralization activity to live SARS-CoV-2 WA-2020 strain in relation to all anti-S immunoglobulin serotypes and IgG subclasses in patients with PAD after SARS-CoV-2 vaccination. The levels of anti-S IgG and IgG1 responses to the vaccine correlated with neutralization activity to the SARS-CoV-2 WA 1/2020 strain in vitro, supporting the implication of anti-S IgG and IgG1 in neutralizing SARS-CoV-2 virus in SARS-CoV-2-vaccinated individuals. This finding also suggests that patients with selective IgG2D likely produce intact humoral immune response to COVID-19 mRNA vaccine.
We assessed cellular immune responses to COVID-19 mRNA vaccines in patients with PAD by measuring EM CD4+ T cells expressing OX40 and 4-1BB in PBMCs stimulated with SARS-CoV-2 S peptides. OX40 and 4-1BB are members of the tumor necrosis factor receptor (TNFR) family, which has a key role in promoting T-cell clonal expansion and immune responses [25, 28, 29]. These molecules have been utilized as markers for CD4+ T-cell activation in patients with COVID-19 [25, 28, 29]. Following the 1st and 2nd doses of COVID-19 mRNA vaccines, the pattern of S-specific CD4+ T-cell response as determined by the frequency of OX40+4-1BB+ CD4+ T cells was similar to that of anti-S IgG in both HC and patients with PADs, suggesting the interface of CD4+ T and B cells in developing both cellular and humoral immunogenicity to the vaccine. Many patients with PADs also present with non-infectious complications including autoimmune disorders and malignancies, and suppression and/or dysregulation of B and T cells are described in PADs [3, 13, 30, 31]. Protection through immunizations is achieved by developing an orchestrated immune response in different cellular subsets including B and T cells, a process that has been shown to be either suppressed or dysregulated in PADs [25, 32]. Therefore, monitoring both humoral and cellular immunity to COVID-19 vaccines in patients with PADs likely provides important information for assessing the development of protective immune responses to the vaccines.
We explored the possible clinical and immunological parameters that predict poor responses to COVID-19 mRNA vaccines in patients with PAD. When responders were defined as having anti-S IgG titers above 0.4 after the 2nd dose of vaccination (based on anti-S IgG titers in HC), all oPAD patients were responders, while 42% of CVID patients were poor responders. The poor responders had significantly lower baseline IgG, IgA, total B, and smB cells compared to the responders, indicating that a fully functional humoral arm of the immune system is essential for the development of anti-S IgG response to COVID-19 mRNA vaccines. Poor responders also had a trend towards increased frequency of EM and EMRA CD8+ T cells compared to responders. EMRA CD8+ T cells are known to have senescent characteristics and expand with aging, chronic infections, autoimmune disorders, and malignancies [33,34,35,36]. In poor vaccine responders, we also noticed an increased frequency of CD21low B cells, a lymphocyte population found to be expanded in CVID patients with autoimmune disorders [37, 38]. Consistent with our findings, Gardulf et al. reported the association of a poor humoral influenza A (H1N1) vaccine response with the increased frequency of memory CD8+ T and CD21low B cells as well as with non-infectious complications including autoimmune manifestations in patients with CVID [39]. Our data thus support that a set of clinical and immune parameters, including history of autoimmune diseases and baseline profile of immunoglobulins, B- and T-cell subsets, could serve as predictive markers for humoral immune responses to COVID-19 mRNA vaccines in patients with CVID.
The limitations of our study include the relatively small numbers of patients with various types of PADs and the lack of long-term follow up of vaccine-specific immune responses. Further investigations of the immunogenicity to COVID-19 mRNA vaccines over the long term in a larger cohort of PAD patients are warranted to properly counsel patients with PADs regarding immune protection against SARS CoV-2 virus during this time of the COVID-19 pandemic.
It has been a challenge for the clinicians to counsel patients with PADs regarding their protection from COVID-19 after administering COVID-19 vaccines because commercially available anti-SARS-CoV-2 spike protein serologic test do not provide protective range of the antibody titer after vaccination. We believe that the results of our study provide new insights into the immunologic response to COVID-19 vaccination and reasonable markers for risk stratification for vaccine immunogenicity in PAD patients which may assist clinicians to identify patients with an increased risk of poor response to COVID-19 vaccines and counsel them accordingly.
In summary, we demonstrated that anti-S IgG, IgA, and IgG subclasses 1 and 3 increased after COVID-19 mRNA vaccination and correlated with neutralization activity against the SARS-CoV-2 WA 1/2020 strain in vitro in HCs and patients with oPADs including IgGD, IgG2D, and sAbD. However, a subset of patients with CVID developed such responses only after the 2nd dose of the vaccines. A similar pattern was also observed by measuring neutralization activity against the SARS-CoV-2 USA-WA 1/2020 strain and S protein-specific CD4+ T-cell responses. The poor vaccine responders in patients with PADs were ones with autoimmune diseases and low levels of baseline IgG, IgG1, IgG2, CD19+ B cells, switched memory B cells and naïve CD8+ T cells, and high level of EM CD8+ T cells.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128.
Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519–33.
Wood P, Stanworth S, Burton J, Jones A, Peckham DG, Green T, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149(3):410–23.
Shields AM, Burns SO, Savic S, Richter AG. COVID-19 in patients with primary and secondary immunodeficiency: the United Kingdom experience. J Allergy Clin Immunol. 2021;147(3):870-5.e1.
Baek MS, Lee M-T, Kim W-Y, Choi JC, Jung S-Y. COVID-19-related outcomes in immunocompromised patients: a nationwide study in Korea. PLoS One. 2021;16(10):e0257641.
Amodio D, Ruggiero A, Sgrulletti M, Pighi C, Cotugno N, Medri C, et al. Humoral and cellular response following vaccination with the BNT162b2 mRNA COVID-19 vaccine in patients affected by primary immunodeficiencies. Front Immunol. 2021;12:727850.
Arroyo-Sanchez D, Cabrera-Marante O, Laguna-Goya R, Almendro-Vazquez P, Carretero O, Gil-Etayo FJ, et al. Immunogenicity of anti-SARS-CoV-2 vaccines in common variable immunodeficiency. J Clin Immunol. 2021;42(2):240–52.
Delmonte OM, Bergerson JRE, Burbelo PD, Durkee-Shock JR, Dobbs K, Bosticardo M, et al. Antibody responses to the SARS-CoV-2 vaccine in individuals with various inborn errors of immunity. J Allergy Clin Immunol. 2021;148(5):1192–7.
Hagin D, Freund T, Navon M, Halperin T, Adir D, Marom R, et al. Immunogenicity of Pfizer-BioNTech COVID-19 vaccine in patients with inborn errors of immunity. J Allergy Clin Immunol. 2021;148(3):739–49.
Squire J, Joshi A. Seroconversion after coronavirus disease 2019 vaccination in patients with immune deficiency. Ann Allergy Asthma Immunol. 2021;127(3):383–4.
Filion CA, Taylor-Black S, Maglione PJ, Radigan L, Cunningham-Rundles C. Differentiation of common variable immunodeficiency from IgG deficiency. J Allergy Clin Immunol Pract. 2019;7(4):1277–84.
Shin JJ, Liauw D, Siddiqui S, Lee J, Chung EJ, Steele R, et al. Immunological and clinical phenotyping in primary antibody deficiencies: a growing disease spectrum. J Clin Immunol. 2020;40(4):592–601.
Shin JJ, Catanzaro J, Yonkof JR, Delmonte O, Sacco K, Shin MS, et al. Infectious complications predict premature CD8(+) T-cell senescence in CD40 ligand-deficient patients. J Clin Immunol. 2021;41(4):795–806.
Guerrera G, Picozza M, D’Orso S, Placido R, Pirronello M, Verdiani A, et al. BNT162b2 vaccination induces durable SARS-CoV-2-specific T cells with a stem cell memory phenotype. Sci Immunol. 2021;6(66):eabl5344.
Reiss S, Baxter AE, Cirelli KM, Dan JM, Morou A, Daigneault A, et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PLoS One. 2017;12(10):e0186998.
Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183(4):996-1012.e19.
Lee N, Shin MS, Kang I. T-cell biology in aging, with a focus on lung disease. J Gerontol A Biol Sci Med Sci. 2012;67(3):254–63.
Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59.
Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174(2):203–11.
Ameratunga R, Longhurst H, Steele R, Lehnert K, Leung E, Brooks AES, et al. Common variable immunodeficiency disorders, T-cell responses to SARS-CoV-2 vaccines, and the risk of chronic COVID-19. J Allergy Clin Immunol Pract. 2021;9(10):3575–83.
Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520.
Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–25.
Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat Immunol. 2021;22(1):67–73.
Klingler J, Weiss S, Itri V, Liu X, Oguntuyo KY, Stevens C et al. Role of IgM and IgA antibodies in the neutralization of SARS-CoV-2. medRxiv. 2020; 23(6):957–70.
Ewer KJ, Barrett JR, Belij-Rammerstorfer S, Sharpe H, Makinson R, Morter R, et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat Med. 2021;27(2):270–8.
Luo H, Jia T, Chen J, Zeng S, Qiu Z, Wu S, et al. The characterization of disease severity associated IgG subclasses response in COVID-19 patients. Front Immunol. 2021;12:632814.
Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68.
Yang L, Liu S, Liu J, Zhang Z, Wan X, Huang B, et al. COVID-19: immunopathogenesis and Immunotherapeutics. Signal Transduct Target Ther. 2020;5(1):128.
Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178(6):3932–43.
Wong GK, Huissoon AP. T-cell abnormalities in common variable immunodeficiency: the hidden defect. J Clin Pathol. 2016;69(8):672–6.
Rodda LB, Netland J, Shehata L, Pruner KB, Morawski PA, Thouvenel CD, et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. Cell. 2021;184(1):169-83.e17.
Kim HR, Hong MS, Dan JM, Kang I. Altered IL-7Ralpha expression with aging and the potential implications of IL-7 therapy on CD8+ T-cell immune responses. Blood. 2006;107(7):2855–62.
Kim JS, Cho BA, Sim JH, Shah K, Woo CM, Lee EB, et al. IL-7Ralphalow memory CD8+ T cells are significantly elevated in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2012;51(9):1587–94.
Park HJ, Shin MS, Kim M, Bilsborrow JB, Mohanty S, Montgomery RR, et al. Transcriptomic analysis of human IL-7 receptor alpha (low) and (high) effector memory CD8(+) T cells reveals an age-associated signature linked to influenza vaccine response in older adults. Aging Cell. 2019;18(4):e12960.
Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144(4):601–13.
Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115(24):5026–36.
Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85.
Gardulf A, Abolhassani H, Gustafson R, Eriksson LE, Hammarstrom L. Predictive markers for humoral influenza vaccine response in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2018;142(6):1922-31.e2.
Funding
This work was supported by funding from the National Institutes of Health (NIH) (1R01AG056728 and R01AG055362 to IK, KL2 TR001862 to JJS, and T32AR07107 to JPY), Quest Diagnostics (to IK and RB), and Burroughs Wellcome Fund (to CBW).
Author information
Authors and Affiliations
Contributions
JJS performed the experiments and chart review, designed the study, analyzed and interpreted the results, and participated in writing the manuscript. JP-Y performed the experiments and chart review, analyzed and interpreted the results, and participated in writing the manuscript. SU and AM performed experiments, analyzed and interpreted the results, and participated in writing the manuscript. H-JP, MSS, RG, and HD designed the study, analyzed and interpreted the results, and participated in writing the manuscript. YA, EZ, JK, AR, FIH, and JC performed chart review, analyzed and interpreted the results, and participated in writing the manuscript. MR, CW, and MM analyzed and interpreted the results and participated in writing the manuscript. RB and IK designed the study, analyzed and interpreted the results, participated in writing the manuscript, and supervised the research.
Corresponding author
Ethics declarations
Ethics Approval
The study was reviewed and approved by the Yale University Institutional Review Board (IRB).
Consent to Participate
The Informed consent for the study was obtained from all participating study subjects.
Consent for Publication
All study subjects were de-identified. No data were associated with personal identifiable information.
Competing Interests
MKR is an employee of Quest Diagnostics. IK and RB received research funding from Quest Diagnostics.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shin, J.J., Par-Young, J., Unlu, S. et al. Defining Clinical and Immunological Predictors of Poor Immune Responses to COVID-19 mRNA Vaccines in Patients with Primary Antibody Deficiency. J Clin Immunol 42, 1137–1150 (2022). https://doi.org/10.1007/s10875-022-01296-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01296-4