
Abstract
Purpose
Chronic granulomatous disorder (CGD) is a primary immunodeficiency which is frequently complicated by inflammatory colitis and is associated with systemic inflammation. Herein, we aimed to investigate the role of the microbiome in the pathogenesis of colitis and systemic inflammation.
Methods
We performed 16S rDNA sequencing on mucosal biopsy samples from each segment of 10 CGD patients’ colons and conducted compositional and functional pathway prediction analyses.
Results
The microbiota in samples from colitis patients demonstrated reduced taxonomic alpha-diversity compared to unaffected patients, even in apparently normal bowel segments. Functional pathway richness was similar between the colitic and non-colitic mucosa, although metabolic pathways involved in butyrate biosynthesis or utilization were enriched in patients with colitis and correlated positively with fecal calprotectin levels. One patient with very severe colitis was dominated by Enterococcus spp., while among other patients Bacteroides spp. abundance correlated with colitis severity measured by fecal calprotectin and an endoscopic severity score. In contrast, Blautia abundance is associated with low severity scores and mucosal health. Several taxa and functional pathways correlated with concentrations of inflammatory cytokines in blood but not with colitis severity. Notably, dividing patients into “high” and “low” systemic inflammation groups demonstrated clearer separation than on the basis of colitis status in beta-diversity analyses.
Conclusion
The microbiome is abnormal in CGD-associated colitis and altered functional characteristics probably contribute to pathogenesis. Furthermore, the relationship between the mucosal microbiome and systemic inflammation, independent of colitis status, implies that the microbiome in CGD can influence the inflammatory phenotype of the condition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic granulomatous disorder (CGD) is a primary immunodeficiency characterized by failure of phagocyte oxidative burst [1]. In addition to life-threatening infection, affected patients frequently suffer inflammatory colitis [2], characterized by cryptitis, crypt abscesses, and crypt architectural distortion as well as granulomas. It is known that the microbiome is altered in other inflammatory bowel diseases and that this may have a causative role in the pathogenesis [3]; this may be of especial importance in CGD where poor innate control of bacteria is a core feature. An important role for the microbiome has been suggested in a mouse model of CGD [4] and the stool microbiota has been described as abnormal in patients with CGD [5].
Microbial dysbiosis is also strongly associated with systemic inflammation of any cause, even in the absence of overt gut disease [6,7,8]. In a recent study, we demonstrated that CGD patients have elevated blood inflammatory markers and cytokines which do not necessarily correlate with the extent of colitis [9].
We therefore first hypothesized that the mucosa-associated microbiome and microbial metabolic pathways would differ between CGD patients with and without colitis, supporting a causative role in the development of colitis as seen in other inflammatory bowel diseases. Next, we hypothesized that the microbiome and microbial pathways would differ according to the extent of systemic inflammation regardless of the extent of colitis, implicating the microbiota in the wider inflammatory phenotype of CGD.
In our recent study, where we demonstrated that CGD-associated colitis can be monitored non-invasively [9], ten participants underwent colonoscopy with biopsies taken from each segment of the large bowel. The blood was also assayed for markers of systemic inflammation. We have here investigated the microbiota in each of these bowel segments and correlated this with the severity of the patients’ colitis and systemic inflammation.
Methods
Patient characteristics have been described previously [9] and are provided in Table 1 and Supplementary Table S1. All patients were receiving antibiotic and antifungal prophylaxis. Colonoscopy and biopsies were performed as part of the study, although there was an urgent clinical indication in one patient with new onset (several weeks) of colitis symptoms; the blood was taken contemporaneously for serum cytokine analysis. Cytokines including interleukin (IL)-1β, IL-6 and IL-12, tumor necrosis factor (TNFα), and a marker of immune activation (soluble cluster of differentiation 14—sCD14) were measured via Luminex technology at the Multiplex Core Laboratory, UMC Utrecht, Netherlands.
A rank scoring was introduced to divide patients into two groups based on cytokine profiles. Briefly, each of the 5 cytokines was scored between 1 and 9 based on the concentration, and the sum of these scores represented a total rank score for each patient. The patients were then split into two groups: (1) high level of systemic inflammation (high—below median total rank score) and (2) low level of systemic inflammation (low—less than or equal to median total rank score). Rank scores and serum IL-1β, IL-6, TNFα, IL-12, and sCD14 measurements are given in Supplementary Table S1. Serum cytokine measurements were not completed for patient P04. Colonic biopsy specimens were obtained from each bowel segment reached (rectum, sigmoid, splenic flexure, hepatic flexure, caecum, terminal ileum), and the presence of colitis in each segment was assessed by the endoscopist and scored according to the Ulcerative Colitis Endoscopic Index of Severity (UCEIS) score. Samples were stored at – 80 °C in RNALater (ThermoFisher).
Metagenomic DNA was extracted from approximately 1 × 1-mm biopsy sections or a blank sample (extraction control) using the DNeasy PowerLyzer PowerSoil kit [10]. For 16S rDNA sequencing, the V3–V4 hypervariable region of the 16S rRNA gene was amplified by PCR using universal 341F and 805R primers fused with Nextera XT index and MiSeq adapter sequences. Molecular grade water was used as a negative control and Mock Community B (HM-783D, www.beiresources.org) was used as a positive control, and amplicons were confirmed on a 1% agarose gel. The extraction kit control and PCR-negative controls did not generate any amplicons, therefore were not included in library pooling. Subsequently, 62 samples including the mock community were pooled at equimolar concentrations and the library was sequenced using a MiSeq sequencer with a 2 × 250-bp paired-end run (Illumina MiSeq, v2 kit). The resulting sequence data was processed using “Quantitative insights into microbial ecology 2” (QIIME2 version 2020.2, https://qiime2.org/) [11]. The raw sequences were de-multiplexed and de-noised using the DADA2 algorithm with default parameters to create amplicon sequence variants (ASVs). The mean sequencing depth was 63,509 (range of 9476–124,026), and the resulting ASVs were assigned taxonomy using the SILVA v132 16S database. Functional metabolic predictions were calculated on ASVs using the PICRUST2 (v2.3.0-b) software with default parameters [12], and the resulting pathway functional profiles were imported into the QIIME2 environment. Taxonomic profiles were generated using the 20 most abundant genera across all samples. Alpha-diversity was calculated on ASVs and predicted functional pathways using the observed ASV index (number of unique features) and Shannon index. For group-wise comparisons at the community level, principal coordinate analysis (PCoA) was performed using the Aitchison distance [13]. The effect of active colitis, history of colitis, immunosuppression, bowel segment, age, CGD type, and individuality was tested by the Adonis test. The ASVs and functional pathway data sets were further standardized by analyzing just two segments (sigmoid and rectum) that were available for all patients. The Mann–Whitney test was used to compare the alpha-diversity metrics between active colitis (AC) and no active colitis (nAC) groups as well as between high and low systemic inflammation groups. Similarly, Aitchison distances were tested for the same groupings using PERMANOVA with 999 permutations for changes in the community composition. Genus level associations for colitis status were investigated using q2-geneiss and q2-ANCOM plugins [14]. Functional pathway data were further analyzed using the DEICODE plugin [15]. The resulting robust Aitchison distances were visualized using PCoA limited to 2 axes and statistical significance was tested using PERMANOVA on the basis of colitis status and systemic inflammation group (High versus Low).
Correlations between fecal calprotectin and genus level taxonomy and functional pathways were calculated using Spearman correlation on the standardized data sets (containing sigmoid and rectum segments). To explore associations between the gut microbiome and inflammatory markers a correlation analysis was performed between the top 20 genera and cytokine concentrations using Spearman’s rank correlation. The top 40 pathways that were associated with the separation of the high and low systemic inflammation groups on axis-1 in the PCoA were further investigated using correlation analyses with cytokine concentrations. Each of the resulting correlation matrices was clustered via hierarchical clustering using Ward’s minimum variance method (Ward.D) and Rho (r2) was reported. Patient P10 was excluded from some analyses, as indicated in the relevant sections, due to the extreme difference in microbial composition from other patients.
Results
The Microbiota of CGD Patients with and Without Colitis Differs in Terms of Dominant Taxa, Alpha-Diversity and Beta-Diversity
Consistent with existing studies, the mucosal microbiome composition showed strong inter-individuality and the differences along the bowel segments within individuals were less than the differences between individuals. Examining the dominant taxa (Fig. 1A), a patient with severe acute colitis (rapid onset of symptoms over several weeks with no prior history of bowel disease) and extremely elevated fecal calprotectin (P10) had a microbiota dominated almost exclusively by Enterococcus. Other patients with colitis exhibited predominantly Bacteroides species, and in total, there were nine bacterial genera which distinguished colitis patients from those without colitis (Supplementary Fig. S1A). Analysis using the q2-gneiss tool revealed that increased proportions of Bacteroides, Clostridium innocuum group, Escherichia–Shigella, and Lachnoclostridium were associated with active colitis, while the greater abundance of Blautia, Alistipes, Bifidobacterium, Dorea, and Subdoligranulum were associated with the non-colitic gut. Notably, in colitis patients, there was no clear difference between segments affected or unaffected by disease, unlike some reports in Crohn’s disease [16]. We used a secondary approach (ANCOM test) to identify differentiating genera, and the difference in abundance of Subdoligranulum was the only statistically significant result. The discovery of this genus—in agreement with prior studies—despite the small size of this cohort, may indicate a functional protective role against colitis development and progression [10, 17].

Genus level gut microbiome profiles of CGD patients with a non-colitic or colitic colon. a Taxonomic profiles of microbiota along the gut for each patient are shown, arranged according to increasing concentrations of fecal calprotectin. In addition to overall colitis status, bowel segments with active colitis are highlighted in red. b Significant correlations between the fecal calprotectin level (measured per patient) and UCEIS score (measured per bowel segment) and relative abundance of Bacteroides and Blautia genus. c Correlation between functional pathways and fecal calprotectin level. For correlation analyses, Spearman’s correlation coefficient (rs) is reported. P10 was excluded from the correlation analyses because of the extreme difference in microbiome composition
To further investigate the drivers of the non-colitis and colitis-associated mucosal microbiome, we performed an exploratory multivariate analysis using Aitchison distances on the 16S data. The results showed significant effects of active colitis, history of colitis (HoC), age, CGD type, and use of immunosuppressants (ImS), although the largest explanatory factor was patient individuality accounting for 33% of the variance (Fig. 2C). The effect of active colitis was evident in 16S alpha-diversity measures in which patients with colitis (n = 5, excluding P10 with almost exclusively Enterococcus) demonstrated reduced taxonomic alpha-diversity compared to those without colitis (n = 4; Fig. 2A). A difference in Shannon index was also seen between those with a history of colitis versus those with no prior colitis (Supplementary Fig. S2) which is in agreement with trends reported previously [5]. Thus, from both alpha- and beta-diversity results, it appears that having a history of colitis is a significant underlying factor which appears to have a long-lasting effect on the composition of the gut microbiome. The use of immunosuppressants did not impact the richness and diversity of the mucosal microbiome in this cohort but did make some contribution to beta-diversity. Eight patients were receiving co-trimoxazole prophylaxis and two were on different antibiotic regimes; however, the outlier patient P10 was in the latter group and thus—although they may be an important contributing factor—analysis on the basis of antibiotics would not be informative. All patients received itraconazole as antifungal prophylaxis.

Alpha and beta-diversity of the patient cohort in relation to active colitis and health. a based on 16S rDNA sequencing data and b based on functional pathways predicted by the PICRUST2 software. (i) Richness (number of ASVs) and diversity (Shannon index) in patients with active colitis (AC) versus those with no active colitis (nAC). Data are shown from the standardized data set with two bowel segments (rectum and sigmoid) per patient. Patient P10, who had an enterococci dominated microbiota, is shown with crossed circles. (ii) Community level clustering of the no colitis and active colitis groups; patients with a history of colitis are also indicated. (iii) Multivariate analysis by Adonis on the Aitchison distances. Adonis formula; distance ~ active colitis + immunosuppression + history of colitis + CGD type + age + gender + patient. Statistical significance between groups is reported in supplementary Table S2. Active colitis, n = 6; no active colitis, n = 4. Active C., active colitis; ImS, immunosuppression; HoC, history of colitis
Bacteroides Abundance Positively Correlates While Blautia Abundance Negatively Correlates with Colitis Severity
We proceeded to investigate correlations between the abundance of bacterial genera and colitis severity. Genus Blautia showed a strong negative correlation (r2 = − 0.81, p = 0.008) with the endoscopic score of disease severity (UCEIS), while the genus Bacteroides showed a positive correlation (r2 = 0.70, p = 0.037) with the same measure (Fig. 1B-ii,iii). Disturbances in some of these taxa have been implicated in other inflammatory bowel diseases suggesting similarities in pathogenesis, and the strong associations with these genera suggest they may be useful as indicators of colitis activity or severity in this cohort [3, 7].
In addition to the UCEIS scoring, elevated levels of fecal calprotectin are associated with intestinal inflammation which can be caused by colitis. In our patients, a level of fecal calprotectin (FCP) above 50 μg/g was indicative of active colitis, and the levels showed some correlation with the Bacteroides genus (r2 = 0.50, p < 0.001; Fig. 1B-i).
Analysis of Microbial Functional Pathways Reveals Differences Between CGD Patients with and Without Colitis
Multivariate analysis using Aitchison distances on functional metabolic pathway predictions also showed significant effects of active colitis, history of colitis (HoC), age, CGD type, and use of immunosuppressants (ImS). Notably, the amount of variation explained by active colitis was almost double (at 24%) that observed in the 16S sequencing data (13%). However, individuality remained the largest explanatory factor at 30% (Fig. 2B-iii). In terms of alpha-diversity, only active colitis and having a history of colitis resulted in significant differences on the Shannon index (Supplementary Table S2). Although the mean difference between groups was small (~ 0.1) for both factors, the ANCOM test revealed differentially abundant pathways. Three pathways were significantly enriched in patients with colitis. These were PWY-6590, CENTFERM-PWY, and FAO-PWY, all of which also showed a significant correlation with the FCP levels, as described in the next section (Supplementary Fig. S1B). On the other hand, GLUCARDEG-PWY (the d-glucarate degradation I pathway) had a higher abundance in patients with prior colitis.
Certain Microbial Functional Pathways Correlate with Colitis Severity as Measured by Fecal Calprotectin
Calprotectin is known to chelate metallic ions such as Zn, Mn, Fe, and Cu, and it can therefore act as an inhibitor for metalloenzymes. This could be a potential underlying reason for the enrichment or reduction of certain metabolic capabilities which cannot be directly inferred by taxonomic assignments. To investigate the relations between the level of FCP and functional characteristics of the mucosal microbiome, a correlation analysis was completed. Fourteen functional pathways were found to significantly correlate (p < 0.05) with the FCP level (Fig. 1C). The first cluster (group-1) contained negatively correlated pathways. Among these, DAPLYSINESYN-PWY (l-lysine biosynthesis I) pathway contains the zinc-dependent dapE metalloenzyme, which plausibly could be limited due to calprotectin mediated sequestration [18]. Moreover, this pathway has been previously identified as one of the differentially abundant pathways between healthy and ulcerative colitis patients [10].
The second cluster (group-2) contained three functional pathways that positively correlated with the FCP levels: (1) PWY-6590, the superpathway of Clostridium acetobutylicum acidogenic fermentation pathway; (2) CENTFERM-PWY, the pyruvate fermentation to butanoate pathway; (3) FAO-PWY, the fatty acid β-oxidation I pathway. Notably, all three are either involved in butyrate biosynthesis or utilization of fatty acids such as butyrate. Compared to the non-colitis patients, the butyrate-producing pathways as well as butyrate utilizing pathways were enriched in patients with colitis, particularly in patients P05 and P07. This suggests that reduced butyrate levels in IBD patients could be due to increased microbial utilization of butyrate [19,20,21], although butyrate levels in CGD colitis have not previously been measured.
In the third cluster, which demonstrated a strong positive correlation with FCP, one of the notable pathways was the PWY-6700 (queuosine biosynthesis I) pathway. Queuosine plays an essential role in tRNA modifications, and the gut microbiome is thought to be a major provider of this micronutrient to the host [22, 23]. However, it was shown to be upregulated under Zn limiting conditions, so again increased FCP may potentially be responsible for its increased abundance in active colitis patients [24, 25].
Systemic Inflammatory Markers Correlate with the Abundance of Certain Bacterial Genera
We next sought to investigate the relation between blood inflammatory markers and mucosal microbiome composition. Correlation analysis showed distinct patterns clustered under four groups (Supplementary Fig. S3). Group-1 mostly consisted of positively correlated genera and included the strongest significant association between Lachnoclostridium and IL12 (r2 = 0.83, p = 0.01). This genus also had increased abundance in patients with active colitis in our cohort and has been previously shown to have increased abundance in colitis but not in Crohn’s disease [17].
Group-3 and Group-4 were almost entirely represented by negative correlations while differing in the associated inflammatory markers. The former included mucosal non-colitic Blautia, Alistipes, and Faecalibacterium genera showing significant negative correlations with IL12, sCD14, and IL1β. In the latter, there were significant negative associations between the innate cytokines (IL6, IL1β, and TNFα) and Agathobacter, Bifidobacterium, Anaerostipes, and Lachnospiraceae NK4A136 group. The majority of these genera (Alistipes, Faecalibacterium, Bifidobacterium, and Lachnospiraceae NK4A136 group) is a short-chain fatty acid (SCFAs; acetate, propionate, and butyrate) producing bacteria that can reduce inflammation [26]. In particular, an association between the increased abundance of Faecalibacterium prausnitzii coupled with increased butyrate and decreased levels of sCD14 has previously been reported in HIV-infected individuals [27]. Interestingly, across the top 20 genera, we were not able to identify as many significant positive as negative correlations with the levels of inflammatory markers. In particular, despite the Bacteroides genus showing significant correlations with the FCP levels and the endoscopic UCEIS score, it did not significantly correlate with any of the systemic inflammatory markers. This lack of positive associations also suggested that there might be additional factors other than colitis which mediate the interplay between the mucosal microbiome and systemic inflammation.
Microbial Composition and Functional Pathway Alpha-Diversity Metrics Differ Between Patients with High and Low Levels of Systemic Inflammation
To explore this possibility, we introduced high (n = 4) and low (n = 4) systemic inflammation groups based on the total rank scores of the inflammatory markers (Supplementary Table S1). Notably, a patient with no colitis and one with the mildest disease were classified into the “high” inflammation group while two patients with active colitis were found to have “low” ranks of inflammatory markers, implying a lack of clear correlation between colonic and systemic inflammation.
Revisiting alpha-diversity metrics, both taxonomic and functional pathway richness showed significant differences between the high and low systemic inflammation groups (Supplementary Table S2). More importantly, inflammation was the only factor that resulted in a significant separation of the functional pathway richness. The low systemic inflammation group had a richness mean of 282 (± 3) unique functional pathways compared to 261 (± 17) in the high inflammation group. Together, these results suggest either that increased systemic inflammation is reducing functional pathway richness in the gut microbiome or conversely (and more plausibly) that reduced functional richness due to disruption of the gut microbiome might be inducing inflammation.
Multivariate Analysis Reveals a Strong Association Between Systemic Inflammation and the Gut Microbiome
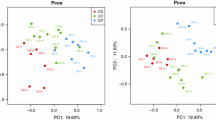
To further investigate the association between gut mucosal microbiome and systemic inflammation, we performed robust Aitchison PCA limited to two dimensions. The initial multivariate analyses showed active colitis as a significant factor explaining 22% and 21% of the variation in the 16S rDNA and functional pathway data, respectively (Supplementary Table S3). However, the greatest explanatory factor was systemic inflammation accounting for 37% of the variation for 16S rDNA and 59% for functional pathway data, even greater than the inter-individual differences. The use of immunosuppressants, CGD type, and history of colitis had more modest effects. The relationship between systemic inflammation and functional and compositional characteristics was further confirmed by PERMANOVA showing a significant and large effect size for both data types (Fig. 3A, B).

Functional pathway and taxonomic associations between mucosal microbiome and systemic inflammation. PCoA of robust Aitchison distances on the basis of systemic inflammation using a ASVs from 16S rDNA sequencing and b functional pathway predictions. Samples with active colitis are shown as circles (○) and no active colitis as squares (□), while fill colors represent high or low systemic inflammation. c Correlation analysis between the top 20 loadings (for both high and low systemic inflammation groups) on axis 1 for functional pathways and inflammatory markers. Significance was tested by PERMANOVA as detailed in Supplementary Table S4
Subsequently, the ANCOM test revealed one genus and five pathways which were differentially abundant between the high and low systemic inflammation groups (Supplementary Fig. S4). The single genus that differed between the systemic inflammation groups was the Lachnospiraceae NK4A136 group, which was completely absent in the patients with high systemic inflammation. This group has been shown to be health associated in various studies [28, 29], as well as considered to have anti-inflammatory properties since it is a SCFA producer [30].
We also sought to investigate whether there was any overlap between the genera associated with colitis and systemic inflammation (Fig. 4). Blautia, Alistipes, and Bifidobacterium were exclusively found to differentiate patients with both non-colitic colon and low systemic inflammation, whereas Lachnoclostridium was exclusively observed as a significant feature in both active colitis and high systemic inflammation. However, five further taxa differentiated colitis from non-colitis without being implicated in systemic inflammation, while six taxa were differentially abundant between the high and low inflammation groups without varying according to colitis status. Again, this implies that the relationship between the gut mucosal microbiome and systemic inflammation is more complex than simply reflecting the severity of colitis.

Genus level taxonomic associations shared across active colitis and systemic inflammation groupings. The Venn diagram concatenates those genera that were identified as significantly different between groups in previous analyses. Lachnoclostridium was shared between colitis and high levels of systemic inflammation, while Blautia, Alistipes, and Bifidobacterium were associated with non-colitic colon and low level of systemic inflammation. C., colitis; Inf., inflammation
Three of the functional pathways, P562-PWY (myo-inositol degradation I), PWY-5304 (superpathway of sulfur oxidation), and P461-PWY (hexitol fermentation to lactate, formate, ethanol, and acetate), had increased abundance in the low inflammation group. Myo-inositol is abundant in the gut, and a higher abundance of its degradation pathway in low systemic inflammation may reflect the presence of a non-dysbiotic mucosal microbiome. PWY-5304 is related to sulfur metabolism in which hydrogen sulfide, l-cysteine, and inorganic sulfate can be produced by microorganisms such as Desulfovibrio species that may have immune-regulatory properties [31,32,33]. Lastly, P461-PWY was found to be more abundant in the low inflammation group. This pathway is responsible for the fermentation of sugar alcohols and has been associated with Anaerostipes hadrus [34].
By contrast, the remaining two pathways had increased abundance in the high inflammation group, and both were involved in the biosynthesis of vitamin B6 (pyridoxal 5′-phosphate, PLP). These were PWY0-845 (superpathway of PLP biosynthesis and salvage) and PYRIDOXSYN-PWY (PLP biosynthesis I). Gut microbiota is one of the main sources of vitamin B6, though the mechanism by which it affects host-microbiome interplay is not well established as there are conflicting reports regarding its relationship to inflammation [35,36,37,38].
The Abundance of Certain Microbial Metabolic Pathways Correlates with Systemic Inflammatory Markers
To further clarify the relationship between the most implicated microbial pathways and inflammation, we performed correlation analysis between the top 20 contributors to axis-1 in the Aitchison PCA and individual cytokines (Fig. 3C). Primarily, pathways that were associated with a lower level of systemic inflammation clustered in group-1 and group-2, and the majority of the significant interactions (14/18) occurred with IL1β and TNFα. The three pathways (PWY-562, PWY-5304, P461-PWY) that were found to have increased abundance in patients with low systemic inflammation also showed significant negative correlations with one or more of the monocyte-derived cytokines. In particular, P461-PWY negatively correlated with IL6, IL1β, and TNFα, which has not been reported previously. In addition, four methionine biosynthesis pathways, PWY-5347 (superpathway of l-methionine biosynthesis), MET-SAM-PWY (superpathway of S-adenosyl-l-methionine biosynthesis), HOMOSER-METSYN (l-methionine biosynthesis I), and HSERMETANA (l-methionine biosynthesis III), showed negative correlations with inflammatory markers. Similarly, a P562-PWY-related pathway, PWY-7237 (myo-, chiro-, and scillo-inositol degradation), showed a negative correlation with TNFα.
In contrast, group-3 and group-4 mostly consisted of positive correlations, although there was only one significantly correlating pathway (PWY0-1533, methylphosphonate degradation I). This pathway had a strong correlation (r2 = 0.71) with the IL12 level, and its abundance was increased in the high inflammation group compared to the low inflammation group. Notably, this pathway has been reported to be enriched in the dysbiotic gut microbiome of severe acute malnutrition patients with acute diarrhea [39]. It was also notable that the FAO-PWY pathway which was positively associated with increased FCP level and identified to have increased abundance in colitis showed a positive correlation (r2 = 0.59) with levels of IL12.
Discussion
The current study is the first to report on the mucosal microbiome of patients with CGD in relation to colitis and systemic inflammation. Given the rarity of this condition, conducting a large-scale investigation would be challenging, and to our knowledge, there is only one previous study describing the fecal microbiome of 11 patients with this immunodeficiency condition [5]. Although this previous study provided valuable first insights on the gut microbiome in CGD, other groups have demonstrated that the localized mucosal microbiota along the gut can significantly differ from that of the fecal in health as well as disease. Therefore, investigating mucosal microbiota in CGD patients may further help us in understanding the regulatory role of the gut microbiota in relation to both colitis and systemic inflammation.
As revealed by the genus level taxonomic profiles, the intra-individual differences along the gut segments were marginal in most cases. In agreement with some [40,41,42,43] but not all [16] previously reported studies, the differences between the colitic and non-colitic segments within individual patients were also minimal in terms of taxonomic composition and alpha-diversity. On the other hand, the expected inter-individual difference between patients was the greatest explanatory factor in the initial beta-diversity analyses independent from colitis status [44, 45]. However, a number of genera were found to associate with either colitic or non-colitic gut. In particular, our results showed that elevated abundance of Subdoligranulum was indicative of normal gut mucosa. Although this was consistent with other studies suggesting its preventative role in IBD [10, 17, 46], one earlier study linked its reduced abundance in IBD patients to the administration of antibiotics [47]. All patients in our study were on antibiotics, although we were unable to analyze according to the agents received as nearly all were on co-trimoxazole. The one patient with an Enterococcus-dominated microbiome was receiving ciprofloxacin and metronidazole and we cannot exclude that these antibiotics were a contributory factor. For example, although the same pattern was not seen in the other patient on ciprofloxacin (plus doxycycline), metronidazole might theoretically have reduced Bacteroides abundance.
Another interesting finding was the strong negative correlation between Blautia and endoscopic assessment scores, in parallel with its increased abundance in non-colitis patients. A similar association was also shown in a study investigating the role of mucosal microbiota dysbiosis in colitis-related cancer. In this study, the authors reported an increased abundance of Blautia in healthy patients along with a possible anti-inflammatory role [48]. However, this bacterium’s presumably protective role in CGD colitis contrasts with a recent study in which it was associated with the IBD-related microbial network in the gut [49]. Notably, such discrepancies can be attributed to the species level functional differences within the genus Blautia. For example, B. coccoides was shown to have a pro-inflammatory effect while B. luti and B. wexlerae displayed anti-inflammatory effects [50, 51].
We also observed a significant positive correlation between the Bacteroides genus and markers of colitis severity. Interestingly, the characteristics of the members of this genus may differ substantially, even at the species level. For example, some B. fragilis can have anti-inflammatory and protective properties against colitis [10, 52, 53], while the enterotoxigenic B. fragilis can induce inflammation and promote IBD [54, 55].
In a related study conducted by Fiedorova and colleagues [44], the authors emphasize the inconsistencies among studies in identifying certain bacteria associating with gastrointestinal disease severity or health in CVID patient cohorts. While there are several host-related (e.g., genetic makeup and CVID characteristics) and environmental factors (e.g., geographical origin, cohort size, and methodology) that could impact the results of such studies, it could also imply that the taxonomic changes may not have to be consistent because the underlying driver is the total functional metabolic capability of the gut microbiota.
With this approach, i.e., analyzing on the basis of microbial functional pathways, we identified an increased number of significant associations by correlation analyses and abundance testing, which improved the contribution of colitis as an explanatory factor in beta-diversity analyses (albeit patient individuality still remained the main factor).
However, the lack of a clear separation between active colitis and no active colitis groups led to the introduction of systemic inflammation levels as a new variable. Surprisingly, the effect size of the existing explanatory factors (e.g., colitis status) and in particular individuality, changed drastically meaning that systemic inflammation was independent from colitis disease severity as reported previously [40]. A dysbiotic gut microbiota was shown to induce systemic inflammation in mice [56], and our findings suggest that high systemic inflammation in CGD can be associated with the altered gut microbial composition and, especially, functional capability. Collectively, these results strongly implicate the colonic mucosal microbiome in the systemic inflammatory phenotype of CGD, independently of the impact of colitis.
The gut microbiome is continuously shaped by a combination of factors, and it can reveal explicit relationships in diseases such as colitis. Our findings also support the concept that there is not a single universal healthy gut microbiota composition. Host lifestyle factors, genetic background, health or specific diseases, the environment, and aging drive microbiota composition may result in several shifts and alterations over time [57,58,59]. For example, we discovered a modest but significant impact of age and genetic type of CGD (X-linked versus autosomal recessive) on beta-diversity in our cohort. The latter might relate to differences in residual neutrophil function, although we have no clear evidence for this at present. Nevertheless, ultimately the microbiome should reach stable homeostasis with the host in terms of metabolic functional capability. These properties of a “healthy” gut microbiome are crucial for understanding the interaction with the host as well as personalized treatment approaches, particularly in immunocompromised patients.
There are a number of limitations to our study that need to be acknowledged. Firstly, our findings are limited to the changes within a CGD patient cohort and did not include healthy individuals for comparison since recruitment of healthy patients was not possible due to sampling by colonoscopy. Secondly, the functional pathway data was generated using a computational prediction tool and may not completely reflect the true functional profiles. Thirdly, given the numbers of patients, we were unable to study the effect of individual immunosuppressants on the microbiota, but compounds such as 5-ASA might also influence the microbial composition. Lastly, non-bacterial members of the gut microbiota such as fungi may provide additional insights but were not analyzed here.
Conclusions
In this work, we have first demonstrated that patients with CGD-associated colitis exhibit reduced diversity in microbial populations at the level of the gut mucosa and identified bacterial taxa which appear to differentiate between non-colitic and colitic colon; the abundance of Bacteroides appears to correlate positively and Blautia negatively with disease activity. Very severe colitis may be associated with the dominance of a single pathogenic species (e.g., Enterococcus). We have also demonstrated differences in microbial metabolism between patients with and without colitis and identified metabolic pathways which associate with disease severity, possibly due to an interaction with fecal calprotectin. Many of the changes in microbiota appear to persist even with mucosal healing and similar patterns are observed in both affected and unaffected segments of patients’ colons, implying a microbial “risk phenotype” for the development of colitis. It will be interesting to study whether this resolves after a successful hematopoietic stem cell transplant or gene therapy.
We have also demonstrated changes in microbial taxa and metabolism corresponding with systemic inflammation, which is not fully explained by the presence of colitis. Indeed, inflammation appears to show clearer and more significant associations with the gut microbiota than colitis itself. Our data therefore imply that CGD patients’ microbiome may influence the inflammatory phenotype of this disease and this demands further investigation. If confirmed in other cohorts, strategies to modify the gut microbiome (including fecal transplant) should be explored as therapies in CGD.
Data Availability
The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) at NCBI BioProject ID: PRJNA613382.
Code Availability
Not applicable.
References
Roos Di. Chronic granulomatous disease. Br. Med. Bull. Oxford University Press; 2016. p. 50–63.
Huang A, Abbasakoor F, Vaizey CJ. Gastrointestinal manifestations of chronic granulomatous disease. Color. Dis. Colorectal Dis; 2006. p. 637–44.
McIlroy J, Ianiro G, Mukhopadhya I, Hansen R, Hold GL. Review article: the gut microbiome in inflammatory bowel disease—avenues for microbial management. Aliment. Pharmacol. Ther. Blackwell Publishing Ltd; 2018. p. 26–42.
Falcone EL, Abusleme L, Swamydas M, Lionakis MS, Ding L, Hsu AP, et al. Colitis susceptibility in p47phox-/- mice is mediated by the microbiome. Microbiome. BioMed Central Ltd.; 2016;4:13.
Sokol H, Mahlaoui N, Aguilar C, Bach P, Join-Lambert O, Garraffo A, et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J Allergy Clin Immunol. Mosby Inc.; 2019;143:775–778.e6.
Telle-Hansen VH, Holven KB, Ulven SM. Impact of a healthy dietary pattern on gut microbiota and systemic inflammation in humans. Nutrients. MDPI AG; 2018.
Arias M, Cobo M, Jaime-Sánchez P, Pastor J, Marijuan P, Pardo J, et al. Gut microbiota and systemic inflammation changes after bread consumption: The ingredients and the processing influence. J Funct Foods Elsevier Ltd. 2017;32:98–105.
Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ: BMJ Publishing Group; 2018.
Lowe DM, Smith PJ, Moreira F, Workman S, Braggins H, Koukias N, et al. Chronic granulomatous disorder–associated colitis can be accurately evaluated with MRI scans and fecal calprotectin level. J Clin Immunol. Springer New York LLC; 2019;39:494–504.
Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature Nature Publishing Group. 2019;569:655–62.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol Nature Publishing Group. 2019;37:852–7.
Douglas G, Maffei V, Zaneveld J, Yurgel S, Brown J, Taylor C, et al. PICRUSt2: an improved and customizable approach for metagenome inference. PICRUSt2 An Improv extensible approach metagenome inference. Cold Spring Harbor Laboratory; 2019;672295.
Aitchison J, Barceló-Vidal C, Martín-Fernández JA, Pawlowsky-Glahn V. Logratio Analysis and compositional distance. Math Geol Kluwer Academic Publishers-Plenum Publishers. 2000;32:271–5.
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. Taylor & Francis; 2015;26:27663.
Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, et al. A novel sparse compositional technique reveals microbial perturbations. mSystems. American Society for Microbiology; 2019;4.
Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. BMC Microbiol; 2011;11.
Qiu Z, Yang H, Rong L, Ding W, Chen J, Zhong L. Targeted metagenome based analyses show gut microbial diversity of inflammatory bowel disease patients. Indian J Microbiol Springer India. 2017;57:307–15.
Gillner DM, Becker DP, Holz RC. Lysine biosynthesis in bacteria: a metallodesuccinylase as a potential antimicrobial target. J. Biol. Inorg. Chem. NIH Public Access; 2013. p. 155–63.
Kumari R, Ahuja V, Paul J. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J Gastroenterol. Baishideng Publishing Group Inc; 2013;19:3404–14.
Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut BMJ Publishing Group. 2014;63:1275–83.
Huda-Faujan N, Abdulamir AS, Fatimah AB, Anas OM, Shuhaimi M, Yazid AM, et al. The impact of the level of the intestinal short chain fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J. Bentham Science Publishers Ltd.; 2010;4:53–8.
Rajakovich LJ, Balskus EP. Metabolic functions of the human gut microbiota: the role of metalloenzymes. Nat. Prod. Rep. Royal Society of Chemistry; 2019. p. 593–625.
Fergus C, Barnes D, Alqasem MA, Kelly VP. The queuine micronutrient: charting a course from microbe to man. Nutrients. MDPI AG; 2015. p. 2897–929.
Pawlik M christin, Hubert K, Joseph B, Claus H, Schoen C, Vogel U. The zinc-responsive regulon of Neisseria meningitidis comprises 17 genes under control of a Zur element. J Bacteriol. American Society for Microbiology Journals; 2012;194:6594–606.
Wang J, Lonergan ZR, Gonzalez-Gutierrez G, Nairn BL, Maxwell CN, Zhang Y, et al. Multi-metal restriction by calprotectin impacts de novo flavin biosynthesis in Acinetobacter baumannii. Cell Chem Biol Elsevier Ltd. 2019;26:745-755.e7.
Venegas DP, De La Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. Frontiers Media S.A.; 2019. p. 277.
Dillon SM, Kibbie J, Lee EJ, Guo K, Santiago ML, Austin GL, et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS Lippincott Williams and Wilkins. 2017;31:511–21.
Li H, Liu F, Lu J, Shi J, Guan J, Yan F, et al. Probiotic Mixture of Lactobacillus plantarum strains improves lipid metabolism and gut microbiota structure in high fat diet-fed mice. Front Microbiol. Frontiers Media S.A.; 2020;11:512.
Stadlbauer V, Engertsberger L, Komarova I, Feldbacher N, Leber B, Pichler G, et al. Dysbiosis, gut barrier dysfunction and inflammation in dementia: a pilot study. BMC Geriatr BioMed Central. 2020;20:1–13.
Zhou Q, Ma L, Zhao W, Zhao W, Han X, Niu J, et al. Flaxseed oil alleviates dextran sulphate sodium-induced ulcerative colitis in rats. J Funct Foods. Elsevier Ltd; 2020;64:103602.
Huisingh J, McNeill JJ, Matrone G. Sulfate reduction by a desulfovibrio species isolated from sheep Rumen1. Appl Microbiol American Society for Microbiology. 1974;28:489–97.
Kushkevych I, Cejnar J, Treml J, Dordević D, Kollar P, Vítězová M. Recent advances in netabolic pathways of sulfate reduction in intestinal bacteria. Cells. MDPI AG; 2020;9:698.
Guo FF, Yu TC, Hong J, Fang JY. Emerging roles of hydrogen sulfide in inflammatory and neoplastic colonic diseases. Front. Physiol. Frontiers Media S.A.; 2016.
Vich Vila A, Imhann F, Collij V, Jankipersadsing SA, Gurry T, Mujagic Z, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018.
Selhub J, Byun A, Liu Z, Mason JB, Bronson RT, Crott JW. Dietary vitamin B6 intake modulates colonic inflammation in the IL10-/- model of inflammatory bowel disease. J Nutr Biochem NIH Public Access. 2013;24:2138–43.
Saibeni S, Cattaneo M, Vecchi M, Zighetti ML, Lecchi A, Lombardi R, et al. Low vitamin B6 plasma levels, a risk factor for thrombosis, in inflammatory bowel disease: role of inflammation and correlation with acute phase reactants. Am J Gastroenterol Am J Gastroenterol. 2003;98:112–7.
Morris MS, Sakakeeny L, Jacques PF, Picciano MF, Selhub J. Vitamin B-6 intake is inversely related to, and the requirement is affected by, inflammation status. J Nutr American Society for Nutrition. 2010;140:103–10.
Huang SC, Wei JCC, Wu DJ, Huang YC. Vitamin B6 supplementation improves pro-inflammatory responses in patients with rheumatoid arthritis. Eur J Clin Nutr. Nature Publishing Group; 2010;64:1007–13.
Kieser S, Sarker SA, Berger B, Sultana S, Chisti MJ, Islam SB, et al. Antibiotic treatment leads to fecal Escherichia coli and coliphage expansion in severely malnourished diarrhea patients. CMGH. Elsevier Inc; 2018;5:458–460.e6.
Lo Presti A, Zorzi F, Del Chierico F, Altomare A, Cocca S, Avola A, et al. Fecal and mucosal microbiota profiling in irritable bowel syndrome and inflammatory bowel disease. Front Microbiol. Frontiers Media S.A.; 2019;10:1655.
Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol Microbiology Society. 2006;55:1141–9.
Sokol H, Lepage P, Seksik P, Doré J, Marteau P. Molecular comparison of dominant microbiota associated with injured versus healthy mucosa in ulcerative colitis [2]. Gut. BMJ Publishing Group; 2007. p. 152–4.
Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen Van Zanten SJO. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. American Society for Microbiology Journals; 2006;44:4136–41.
Fiedorová K, Radvanský M, Bosák J, Grombiříková H, Němcová E, Králíčková P, et al. Bacterial but not fungal gut microbiota alterations are associated with common variable immunodeficiency (CVID) phenotype. Front Immunol. Frontiers Media S.A.; 2019;10:1914.
Dill-McFarland KA, Tang ZZ, Kemis JH, Kerby RL, Chen G, Palloni A, et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. Nature Publishing Group; 2019;9.
Kaakoush NO, Day AS, Huinao KD, Leach ST, Lemberg DA, Dowd SE, et al. Microbial dysbiosis in pediatric patients with Crohn’s disease. 2012;
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward D V., et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. BioMed Central; 2012;13:R79.
Richard ML, Liguori G, Lamas B, Brandi G, Costa G da, Hoffmann TW, et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes. Taylor & Francis; 2018;9:131.
Yilmaz B, Juillerat P, Øyås O, Ramon C, Bravo FD, Franc Y, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med Nature Publishing Group. 2019;25:323–36.
Tuovinen E, Keto J, Nikkilä J, Mättö J, Lähteenmäki K. Cytokine response of human mononuclear cells induced by intestinal Clostridium species. Anaerobe Academic Press. 2013;19:70–6.
Benítez-Páez A, Gómez del Pugar EM, López-Almela I, Moya-Pérez Á, Codoñer-Franch P, Sanz Y. Depletion of Blautia species in the microbiota of obese children relates to intestinal inflammation and metabolic phenotype worsening . mSystems. American Society for Microbiology; 2020;5.
Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature Nature Publishing Group. 2008;453:620–5.
Blandford LE, Johnston EL, Sanderson JD, Wade WG, Lax AJ. Promoter orientation of the immunomodulatory Bacteroides fragilis capsular polysaccharide A (PSA) is off in individuals with inflammatory bowel disease (IBD). Gut Microbes. Taylor and Francis Inc.; 2019;10:569–77.
Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe Cell Press. 2018;23:203-214.e5.
Zamani S, Hesam Shariati S, Zali MR, Asadzadeh Aghdaei H, Sarabi Asiabar A, Bokaie S, et al. Detection of enterotoxigenic Bacteroides fragilis in patients with ulcerative colitis. Gut Pathog. BioMed Central Ltd.; 2017;9.
Brandsma E, Kloosterhuis NJ, Koster M, Dekker DC, Gijbels MJJ, Van Der Velden S, et al. A proinflammatory gut microbiota increases systemic inflammation and accelerates atherosclerosis. Circ Res Lippincott Williams and Wilkins. 2019;124:94–100.
Xu F, Fu Y, Sun TY, Jiang Z, Miao Z, Shuai M, et al. The interplay between host genetics and the gut microbiome reveals common and distinct microbiome features for complex human diseases. Microbiome. BioMed Central Ltd; 2020;8:145.
van Schewick CM, Nöltner C, Abel S, Burns SO, Workman S, Symes A, et al. Altered microbiota, impaired quality of life, malabsorption, infection, and inflammation in CVID patients with diarrhoea. Front Immunol. Frontiers Media S.A.; 2020;11:1654.
Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BAH, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature Nature Research. 2016;535:376–81.
Funding
Rare Disease Foundation/BC Children’s Hospital Foundation (Grants 1915 and 1920 to D.M.L.) and UCLH Biomedical Research Centre (BRC299/III/DL/101350 to D.M.L.).
Author information
Authors and Affiliations
Contributions
M.D. performed laboratory and bioinformatic analyses. S.H. collected clinical data. P.J.S. and C.D.M. helped to conceive the study, performed endoscopy with assessment of colitis activity, and obtained biopsies. D.M.L. conceived the study, recruited patients, and supervised the analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
NHS Research Ethics Committee (REC) 15/LO/1334.
Consent to Participate
All patients provided written informed consent.
Consent for Publication
All participants consented to the results of the study to be published. No individual details, images, or videos are included in this manuscript.
Competing Interests
D.M.L. has received travel and subsistence costs for consultancy work for CSL Behring and fees for a roundtable discussion from Merck. Other authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Davrandi, M., Harris, S., Smith, P.J. et al. The Relationship Between Mucosal Microbiota, Colitis, and Systemic Inflammation in Chronic Granulomatous Disorder. J Clin Immunol 42, 312–324 (2022). https://doi.org/10.1007/s10875-021-01165-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-021-01165-6