
Abstract
Purpose
The ubiquitous calcium–independent phospholipase A2 enzyme (iPLA2) is inhibited by calmodulin binding and known to be responsible for phospholipid remodeling housekeeping functions including granule exocytosis–associated membrane fusion in normal human neutrophils. We evaluate in human neutrophils the iPLA2 secretagogue effects using normal neutrophils, where reactive oxygen species (ROS) generation has been blocked by diphenyleneiodonium, as well as in neutrophils from chronic granulomatous disease (CGD) patients.
Methods
Neutrophils were pretreated with W7, a calmodulin inhibitor known to activate iPLA2 and exocytosis of granules, and vesicles as well as intra- and extra-microbicidal activity against Staphylococcus aureus and Aspergillus fumigatus were evaluated.
Results
W7 increases exocytosis of primary, secondary, and tertiary granules and vesicles and improves neutrophil microbicidal activity against S. aureus and A. fumigatus.
Conclusions
In neutrophils, calmodulin-mediated iPLA2 inhibition controls granule and vesicle exocytosis in the phagosome and in the extracellular microenvironment. Relieving iPLA2 inhibition results in increased exocytosis of primary, secondary, and tertiary granules and secretory vesicles with correction of defective intracellular and extracellular microbicidal activity. In CGD patients presenting ROS defective production, this increase in the non-oxidative killing pathway partially corrects their microbicidal defects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polymorphonuclear cells (PMN) kill micro-organisms by oxygen-dependent and oxygen-independent mechanisms. Stimulation of neutrophil plasma membrane NADPH oxidase by a number of stimuli leads to the production of reactive oxygen species (ROS) directly toxic to the micro-organisms. Oxygen-independent killing depends on the delivery either into the phagosome or into the extracellular environment of pre-formed anti-microbial compounds stored in the neutrophil granules and vesicles. Inherited disorders with an inability to kill certain types of bacteria and/or fungus leading to recurrent life–threatening infections are seen in a variety of rare diseases: chronic granulomatous disease (CGD) patients have an inherited functional defect of the NADPH oxidase complex associated with absent or greatly reduced production of superoxide anion and downstream ROS; in contrast, Chediak-Higashi patients or patients having neutrophil-specific granule deficiency have an intact NADPH oxidase activity but defective granule mobilization or absent granules. Controversy exists whether ROS or the anti-microbial peptides are the neutrophil’s microbe-killing machines [1]. On the other hand, collaboration within the neutrophil among oxidants and granule proteins may be necessary for optimal microbe damage [2]. Thus, among Gram-positive micro-organisms, individuals with CGD have recurrent Staphylococcus aureus (S. aureus) infections which require oxidative mechanisms for efficient killing while they do control Streptococcus pneumoniae infections through effective exocytosis of a number of granule-derived microbicidal proteins [3].
Phospholipase A2 (PLA2) enzymes act on membrane phospholipids generating lipid metabolites that have an essential role in the final stage of membrane fusion [4]. The calcium-independent PLA2 (iPLA2) enzyme present in terminally differentiated granulocytes has been associated with cell division and granule exocytosis–associated membrane fusion [4,5,6]. In the cytosol, iPLA2 activity is inhibited through calmodulin binding and in human PMN, exposure to molecules containing calmodulin binding motifs results in increased in iPLA2 activity together with exocytosis of anti-microbial peptides [6].
To further evaluate the iPLA2 secretagogue effect in myeloid cell physiology, we investigated in both normal human PMN and in PMN where ROS generation was blocked by DPI how iPLA2 activation through W7-induced calmodulin inhibition was associated with (i) upregulation of primary, secondary, and tertiary granules and secretory vesicles, (ii) increased microbicidal activity, and (iii) compared these results with those obtained after cell exposure to the powerful PMN-activating chemotactic factor fMLP. We also assessed the role of iPLA2 activation in PMN isolated from CGD patients.
Materials and Methods
Isolation of Human Neutrophils and Reagents
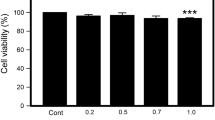
Neutrophils were isolated from citrated venous blood of healthy volunteers using a one-step separation Polymorphoprep (Axis-Shield). When indicated, cells were pretreated with the calmodulin antagonist W7 (50 μM) or its less active congener W12 (50 μM), for 60 min, with bromoenol lactone (BEL), 10 μM, a suicide substrate inhibitor of iPLA2, for 30 min, with the R- or S-isomer of BEL (R- and S-BEL), 25 μM for 30 min or with DPI (10 μM) for 15 min, a concentration which does not affect viability or phagocytosis and inhibit by more than 95% ROS production [7]. Cell viability after W7 treatment was assessed by their ability to mobilize calcium by platelet-activating factor (PAF) or fMLP (Supplementary Figure S1).
Patient Characteristics
PMN were obtained from 9 CGD patients after written authorization in accordance with Declaration of Helsinki, following protocol approved by the Hôpital des Enfants ethical committee. Genotyping of 9 CGD patients revealed a group of 5 males with gp91 phox–linked disease, 2 gp91 phox heterozygous females presenting multiple infections and in vitro defect in ROS production (30% and 5% using flow cytometry based DCFH), and 1 female with autosomal p67 phox deficiency. Homozygous autosomal recessive CGD was diagnosed in one female patient without conclusive genotyping based on the absence of ROS production (0%) by PMN using flow cytometry–based DCFH assay.
Degranulation Assays
Cell surface expression of CD63, CD66b, and CD35 was monitored by flow cytometry for assessing degranulation of azurophilic and specific granules and secretory vesicles respectively [8]. PMN were either untreated or pretreated with DPI (10 μM) for 15 min or BEL (10 μM) for 30 min, R- or S-BEL (25 μM) for 30 min, followed by stimulation with W7 (50 μM) or W12 (50 μM) for 60 min or fMLP (10−6 M) for 20 min (CD63) or 60 min (CD66b, CD35) before staining with FITC-coupled antibodies for 20 min at 4 °C. Lysis solution (BD Biosciences) was used to lyse residual red blood cells and to fix the cells before flow cytometry analysis. The release in the supernatant of α defensin, lactoferrin, and MMP-9 was monitored for assessing degranulation of azurophilic and specific and gelatinase granules respectively in cells (1 × 107/ml) pretreated or not with cytochalasin B (Cyt B, 5 μg/ml) for 20 min and exposed to W7 (50 μM) for 60 min. Released α-defensin and lactoferrin (Hycult Biotech) and MMP-9 (R and D systems) activity were measured by ELISA according to the manufacturer’s recommendations. The release in the supernatant of MPO, a marker of azurophilic granules, was monitored in cells pretreated or not with CB and exposed to W7 (50 μM) for 60 min or fMLP (10−6 M) for 10 min. Released MPO activity was measured using a chromogenic substrate as described by the manufacturer (Sigma).
iPLA2 Activity Assay
PMN (1 × 107 cells/ml) were collected, sonicated 3 times for 10 s with a 15-s break between cycles, and centrifuged at 15,000g for 20 min at 4 °C. The supernatant was removed and kept on ice. After determination of the protein concentration (Bradford assay), iPLA2 activity measurement was obtained in presence of W7 and R-BEL using the cPLA2 assay kit (Cayman chemical) [6]. Activity of iPLA2 is expressed as percent of control (untreated cells). Results were from ten similar experiments, and each was performed in duplicate.
Phagocytosis Assay
Bacterial phagocytosis was evaluated by flow cytometry [9]. PMN were exposed to W7 (50 μM) for 60 min prior to addition of FITC-labeled S. aureus (subsp. aureus ROSENBACH, ATCC 25923) opsonized with serum from the same donor at a ratio of 1:10 at 37 °C. Fluorescence intensity of bacteria was determined after 15 min.
Intracellular and Extracellular S. aureus Killing
For intracellular killing assays [9], cells untreated or pretreated with DPI were exposed to W7 or fMLP. Cells were then counted again and an identical number (1 × 107/ml) was incubated with human serum opsonized (5 min) S. aureus (4 McFarland standard, 1.2 × 109 bacteria suspension/ml) for 20 min. A 100-μL sample was then diluted into 1 ml ice-cold water (pH 11) to lyse neutrophils. Each sample was diluted further into water (pH 7) and spread on nutrient agar plates. Plates were incubated overnight at 37 °C and the number of colonies formed was counted. For extracellular killing assays, supernatant of 1 × 107/ml CB-primed neutrophils exposed to either buffer or W7 of fMLP were incubated with S. aureus for 60 min. The bacteria were spun down, resuspended, plated, and incubated for 18 h and percent S. aureus survival was calculated based on colony plate counting [10]. W7 alone did not affect the S. aureus viability.
Intracellular and Extracellular Killing of Aspergillus
To assess the hyphae killing by granulocytes, a clinical Aspergillus fumigatus isolate was used. Conidia were overnight incubated in HBSS and young hyphae were co-cultured with untreated or treated PMNs (1 × 107/ml) for 20 h [9]. Hereafter, cells were lysed in distilled water (pH 7) and hyphae injury was quantified by measuring the metabolic activity of fungi by reduction of the tetrazolium reagent XTT. To assess non-oxidative hyphae killing, young hyphae were incubated with neutrophil supernatant and cell-free hyphae killing was measured by XTT reduction assay [11]. W7 alone did not affect the hyphae viability.
Statistical Analysis
Data were analyzed as appropriate either by Wilcoxon matched-pairs test or by ANOVA using GraphPad Prism Software v3.0. Results are expressed as mean ± standard error of the mean (SEM). A p value ≤ 0.05 indicated significant differences.
Results and Discussion
Increased membrane expression by flow cytometry of CD63, CD66b, and CD35 was used respectively to quantify exocytosis of primary (azurophilic), secondary (specific) granules, and secretory vesicles [10, 12]. Detection in the supernatant of α-defensin and MPO was used to evaluate exocytosis of primary granules while lactoferrin and MMP-9 measurements were respectively for secondary and tertiary (gelatinase) granule exocytosis [13]. In normal human PMN, W7 increases membrane expression of CD63, CD66b, and CD35 (Fig. 1a–c). W7 alone inhibits ROS production induced by fMLP by more than 95% [14, 15]. Additional inhibition of NADPH oxidase by DPI did not affect the W7 exocytosis increase of azurophilic granules and vesicles (Fig. 1d, f). However, it inhibited the exocytosis of secondary granules (Fig. 1e). BEL, a selective iPLA2 inhibitor, inhibited the exocytosis of azurophilic granules and secretory vesicles (Fig. 1d, f). In contrast to W7, the less active calmodulin antagonist W12, the unchlorinated analogue of W7, did not affect exocytosis (Fig. 1g–j).

The calmodulin inhibitor W7 stimulates granule and vesicle exocytosis. (a, b, and c). Release of subset types of granules and vesicles was tested by flow cytometry following cell surface expression of CD63 (azurophilic granules), CD66b (specific granules), and CD35 (secretory vesicles) in neutrophils treated as described in material and methods. a–c Histogram overlays showing the expression of granule markers (CD63, CD66b, and CD35) on neutrophils following treatment either with W7 (bold lines), fMLP (dotted lines), or with vehicle control (thin lines), representative data from one blood donor is shown. d–f Results are expressed as mean ± SEM between control, W7, and fMLP pretreated cells and exposed to DPI or BEL as described in the “Materials and Methods” section. g–j W12 does not induce cell surface expression of CD63, CD66b, and CD35 as assessed by flow cytometry. g–i Histogram overlays showing the expression of granule markers (CD63, CD66b, and CD35) on neutrophils following treatment either with W7 (bold lines), W12 (dotted lines), or vehicle control (thin lines), representative data from one blood donor is shown. A minimum of 9 independent experiments in duplicate were performed. Results are expressed as mean ± SEM. kR-BEL but Not S-BEL inhibits CD35 upregulation by W7. PMN were pretreated by 10 μM BEL, 25 μM R-BEL, or 25 μM S-BEL for 30 min before addition of W7, CD35 was determined as above. Data are represented as the mean ± S.E.M. of at least six separate experiments. l iPLA2 activity is increased in PMN pretreated with W7. iPLA2 activity after treatment by 50 μM W7 or/and R-BEL 25 μM an iPLA2γ inhibitor was measured as described in the “Materials and Methods” section using control cells as 100% enzyme activity. Results from ten independent experiments are presented as mean ± SEM
The racemic form of BEL used in our experiments is constituted by the 2 enantiomers R-BEL and S-BEL, R-BEL being approximately an order of magnitude more selective for iPLA γ than iPLA2 β [16]. As shown in Fig. 1k, R-BEL but not S-BEL inhibits CD35 upregulation by W7. In human PMN, BEL including R and S-BEL has been reported to inhibit iPLA2 activity [6]; in our study, we confirm that R-BEL is involved in this inhibition, as shown in Fig. 1l.
The cortical actin network prevents granules accessing to the plasma membrane [12]; primary granules which contain the largest number of bactericidal proteins are the most difficult granules to mobilize [13]. However, disruption of the actin cytoskeleton by cytochalasin B was not necessary to observe W7-increased exocytosis of all granule subsets (Fig. 2a) while it was mandatory for fMLP-induced exocytosis of MPO-derived primary granules (Fig. 2b).

W7 triggers the release of α-defensin, lactoferrin, and MMP-9. a Human PMN pretreated or not with cytochalasin B were stimulated with W7 and cell-free supernatants were assayed by ELISA for α-defensin, lacotoferrin, and MMP-9 contents. A minimum of 9 independent experiments in duplicate were performed. Results are expressed as mean ± SEM. b Human PMN pretreated or not with cytocholasin B were stimulated with either W7 or with fMLP and released MPO, a marker of primary granules, was monitored using a chromogenic substrate. Statistics as in (a). c The calmodulin inhibitor W7 corrects defective bactericidal and fungicidal activity in DPI-treated PMN. c Human neutrophils, untreated or pretreated with DPI were exposed to W7 or fMLP as described in the “Materials and Methods” section. Cells were washed, counted, and an identical number of cells were incubated with serum opsonized S. aureus and survival was assessed following a 20-min incubation. On the graph, the numbers of colonies with non stimulated PMN (NS) represent 100% killing. Raw data including number of bacterial colonies are shown in Table S2. For supernatant killing assays, supernatant of 1 × 107/ml untreated, W7 or fMLP treated PMN were incubated with S. aureus for 60 min. The bacteria were spun down, resuspended, plated, and incubated for 18 h and percent S. aureus survival was calculated based on colony plate counting. No difference was observed between NS and DPI pretreated cells (not shown). d To assess the hyphae killing by granulocytes, conidia were overnight incubated in HBSS and young hyphae were co-cultured with untreated or treated PMN (1 × 107/ml) as in A for 20 h. Hereafter, cells were lysed in distilled water (pH 7) and hyphae injury was quantified by measuring the metabolic activity of fungi by reduction of the tetrazolium reagent XTT. To assess non-oxidative hyphae killing, young hyphae were incubated with neutrophil supernatant prepared as in a and hyphae killing was measured by XTT reduction assay. e W7 does not modify human neutrophil phagocytosis as assessed by counting the uptake of opsonized S. aureus as described in the “Materials and Methods” section. A minimum of 9 independent experiments performed in duplicate were performed for bacterial killing. A minimum of 6 independent experiments in duplicate were performed for A. fumigatus killing and phagocytosis. Results are expressed as mean ± standard error of the Mean (SEM)
Based on these results, we hypothesize that in normal PMN, calmodulin-mediated iPLA2 inhibition controls granule and secretory vesicle exocytosis. iPLA2 activity can be released from this tonic inhibition resulting in granule and vesicle exocytosis in the phagosome and into the extracellular microenvironment. To evaluate the microbicidal activity of W7-associated increased exocytosis, both intracellular and extracellular killing against S. aureus was performed using W7-treated PMN where NADPH oxidase activity has been inhibited by DPI. DPI-treated PMN display a profound intracellular bactericidal defect against S. aureus which is partially restored by W7, while fMLP does not have any corrective effect (Fig. 2c). This increased in W7-associated bactericidal activity is also observed in extracellular killing assays (Fig. 2c). Similarly, in DPI-treated PMN, there is a profound defect in intracellular anti-aspergillus activity which is also partially restored by W7 (Fig. 2d). In contrast to fMLP, A. fumigatus supernatant killing was increased in W7-exposed cells (Fig. 2d). Of interest, although W7 inhibits by more than 95% ROS production in PMN [14, 15], there were no statistical differences in either intra- and extra-bactericidal activity or intra- and extra-fungicidal activity between PMN and W7-treated PMN. Since W7 does not have any effect on phagocytosis (Fig. 2e), our results support the hypothesis that the correction of the microbicidal defects in W7-treated cells is associated with secretagogue effects on granules and vesicles and increased exocytosis of microbicidal compounds in the phagosome or into the micro-environment.
In CGD neutrophils, non-oxygen-dependent killing occurs indicating that degranulation and release of microbicidal proteins occur [7, 17]. We then evaluated the activity of W7 on PMN isolated from nine CGD patients. In these cells, W7 increased CD63 and CD35 membrane expression (Fig. 3a, b, c–e) while no effect was seen on CD66b expression (data not shown); this last result obtained from only three patients agrees with our data obtained from normal DPI–treated PMN where W7 exposure did not upregulate CD66b expression (Fig. 1e).

W7 induces degranulation of azurophilic and secretory granules and increases S. aureus killing in CGD neutrophils. a–e Cell surface expression of CD63 (n = 8) and CD35 (n = 9) was monitored by flow cytometry as described in Fig. 1a, c in CGD PMN exposed to W7 (50 μM) for 1 h. f Increased intracellular killing of S. aureus by CGD PMN (n = 7) was determined by colony plate counting as described in Fig. 1g. Cells were treated with buffer or W7 (50 μM) for 1 h before incubation with S. aureus. Early and late killing was measured after 20-min or 30-min incubation with bacteria
In addition, W7 partially corrected the deficit in intracellular bactericidal activity against S. aureus presented by the CGD PMN (Fig. 3f). Thus in CGD patients presenting ROS defective production, this increase in the non-oxidative killing pathway partially corrects their microbicidal defects.
Taken together, our study extends the results obtained previously in human PMN where iPLA2 activation by neuropeptides was associated with secretion of a number of peptides associated with innate immunity [6]. These peptides detected by proteomic analysis were found to have anti-microbial and anti-inflammatory properties and similarly to our results were stored in the 3 types of granules including the primary granules, the most difficult ones to mobilize. The ability of R-BEL but not S-BEL to upregulate CD35 membrane expression and our results indicating that R-BEL activate iPLA2 support the hypothesis that iPLA2γ rather than iPLA2β plays a role in myeloid granular exocytosis.
Besides being the first line of defense against invading pathogens, PMN are also important in the orchestration of adaptive immunity: PMN cross-talk with lymphocytes and antigen-presenting cells via mediators contained in their granules [18]. Thus, modulation of neutrophil exocytosis by calmodulin binding molecules might also be of interest in non-infectious disorders such as inflammation, auto-immunity, and cancer. In this regard, we found that the high affinity IgG binding Fc receptor, FcɣR1 or CD64, contained in the azurophilic granules and involved in tumor cell killing [19] through antibody-dependent cellular cytotoxicity, is also highly upregulated by W7.
W7 as an anti arrhythmic drug has been used successfully in vivo in rodents and dogs without major toxicities [20]. However, W7 is associated with neuronal death in rabbits’ cultured cortical neurons [21]. Thus, more insight into long-term W7 effects is necessary before its use as a potential therapeutic drug. Of interest, potential therapeutic small-molecules inhibitors of iPLA2 enzymes have been developed and are now evaluated in clinical trials in order to treat inflammatory diseases [22].
Abbreviations
- BEL:
-
bromoenol lactone
- Cyt B:
-
cytochalasin B
- CGD:
-
chronic granulomatose disease
- DCFH:
-
dichloro-dihydro-fluorescein
- DPI:
-
diphenyleneiodonium
- iPLA2:
-
calcium-independent phospholipase A2
- MMP-9:
-
matrix metallo-proteinase 9
- MPO:
-
myeloperoxidase
- PAF:
-
platelet-activating factor
- PMN:
-
polymorphonuclear leukocyte
- ROS:
-
reactive oxygen species
- XTT:
-
sodium 3,3′-[1(phenylamino)carbonyl]-3,4-tetrazolium]-3is(4-methoxy-6-nitro) benzene sulfonic acid hydrate
References
Williams R. Killing controversy. J Exp Med. 2006;203(11):2404.
Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102.
Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol. 2009;183(4):2602–9.
Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, Lei X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res. 2015;56(9):1643–68.
Larsson Forsell PK, Runarsson G, Ibrahim M, Björkholm M, Claesson HE. On the expression of cytosolic calcium-independent phospholipase A2 (88kDa) in immature and mature myeloid cells and its role in leukotriene synthesis in human granulocytes. FEBS Lett. 1998;434(3):295–9.
Zhang D, Shooshtarizadeh P, Laventie BJ, Colin DA, Chich JF, Vidic J, et al. Two chromogranin a-derived peptides induce calcium entry in human neutrophils by calmodulin-regulated calcium independent phospholipase A2. PLoS One. 2009;4(2):e4501.
Ellis JA, Mayer SJ, Jones OT. The effect of the NADPH oxidase inhibitor diphenyleneiodonium on aerobic and anaerobic microbicidal activities of human neutrophils. Biochem J. 1988;251(3):887–91.
Simard JC, Girard D, Tessier PA. Induction of neutrophil degranulation by S100A9 via a MAPK-dependent mechanism. J Leukoc Biol. 2010;87(5):905–14.
Stemerding AM, Köhl J, Pandey MK, Kuipers A, Leusen JH, Boross P, et al. Staphylococcus aureus formyl peptide receptor-like 1 inhibitor (FLIPr) and its homologue FLIPr-like are potent FcγR antagonists that inhibit IgG-mediated effector functions. J Immunol. 2013;191(1):353–62.
Flamand L, Tremblay MJ, Borgeat P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J Immunol. 2007;178(12):8036–45.
Lee MJ, Liu H, Barker BM, Snarr BD, Gravelat FN, Al Abdallah Q, et al. The fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog. 2015;11(10):e1005187.
Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR. The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. Am J Physiol Cell Physiol. 2007;292(5):C1690–700.
Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28(8):340–5.
Jian Lian JP, Crossley L, Zhan Q, Huang R, Coffer P, Toker A, et al. Antagonists of calcium fluxes and calmodulin block activation of the p21-activated protein kinases in neutrophils. J Immunol. 2001;166(4):2643–50.
Verploegen S, van Leeuwen CM, van Deutekom HW, Lammers JW, Koenderman L, Coffer PJ. Role of Ca2+/calmodulin regulated signaling pathways in chemoattractant induced neutrophil effector functions. Comparison with the role of phosphotidylinositol-3 kinase. Eur J Biochem. 2002;269(18):4625–34.
Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem. 2002;277:32807–14.
Odell EW, Segal AW. Killing of pathogens associated with chronic granulomatous disease by the non-oxidative microbicidal mechanisms of human neutrophils. J Med Microbiol. 1991;34(3):129–35.
Leliefeld PH, Koenderman L, Pillay J. How neutrophils shape adaptive immune responses. Front Immunol. 2015;6:471.
Bakema JE, Ganzevles SH, Fluitsma DM, Schilham MW, Beelen RH, Valerius T, et al. Targeting FcαRI on polymorphonuclear cells induces tumor cell killing through autophagy. J Immunol. 2011;187(2):726–32.
Driessen HE, Bourgonje VJ, van Veen TA, Vos MA. New antiarrhythmic targets to control intracellular calcium handling. Neth Heart J. 2014;22(5):198–213.
Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, et al. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287:8495–506.
Nikolaou A, Kokotou MG, Vasilakaki S, Kokotos G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim Biophys Acta Mol Cell Biol Lipids. 2019 Jun;1864(6):941–56.
Acknowledgements
The authors thank Drs. Christophe Chantrain, Alice Ferster, Benoit Florkin, Pierre Philippet, and Jutte Van Der Werff Ten Bosch for the CGD blood samples and Drs. Brigitte Cantiniaux and Francis Corazza for the discussion.
Funding
This work was supported by the Kriibskrank Kanner Foundatioun of Luxembourg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
PMN were obtained from 9 CGD patients after written authorization in accordance with Declaration of Helsinki, following protocol approved by the Hôpital des Enfants ethical committee
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 223 kb)
Rights and permissions
About this article

Cite this article
Harfi, I., D’Hondt, S. & Sariban, E. iPLA2 Activation Mediates Granular Exocytosis and Corrects Microbicidal Defects in ROS-Deficient and CGD Human Neutrophils. J Clin Immunol 39, 486–493 (2019). https://doi.org/10.1007/s10875-019-00630-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-019-00630-7