
Abstract
Purpose
X-linked agammagobulinemia (XLA) is a primary immunodeficiency caused by Bruton’s tyrosine kinase (BTK) gene defect. XLA patients have absent or reduced number of peripheral B cells and a profound deficiency in all immunoglobulin isotypes. This multicenter study reports the clinical, immunological and molecular features of Bruton’s disease in 40 North African male patients.
Methods
Fifty male out of 63 (male and female) patients diagnosed with serum agammaglobulinemia and non detectable to less than 2 % peripheral B cells were enrolled. The search for BTK gene mutations was performed for all of them by genomic DNA amplification and Sanger sequencing.
Results
We identified 33 different mutations in the BTK gene in 40 patients including 12 missense mutations, 6 nonsense mutations, 6 splice-site mutations, 5 frameshift, 2 large deletions, one complex mutation and one in-frame deletion. Seventeen of these mutations are novel. This large series shows a lower frequency of XLA among male patients from North Africa with agammaglobulinemia and absent to low B cells compared with other international studies (63.5 % vs 85 %). No strong evidence for genotype-phenotype correlation was observed.
Conclusions
This study adds to other reports from highly consanguineous North African populations, showing lower frequency of X-linked forms as compared to AR forms of the same primary immunodeficiency. Furthermore, a large number of novel BTK mutations were identified and could further help identify carriers for genetic counseling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
X-linked agammaglobulinemia (XLA; MIM# 300300), first described in 1952 [1], represents the prototype for primary immune deficiencies and was among the first monogenic immunological disorders for which a genetic cause has been discovered [2]. XLA phenotype is characterized by dramatically reduced or absent mature B lymphocytes (less than 2 % of total lymphocytes), which is caused by a differentiation blockage affecting the transition of B cell progenitors to mature B lymphocytes [3]. Affected individuals have profound hypogammaglobulinemia and thus show increased susceptibility to severe and recurrent bacterial infections [4]; including infections of the upper and lower respiratory tract and the skin, meningoencephalitis, gastroenteritis and conjunctivitis [5]. Age at onset is usually between 6 and 12 months of age, coinciding with the catabolism of maternal IgG. XLA patients may also develop purulent and non-purulent arthritis, osteomyelitis, and protracted enterovirus infection [5]. The prognosis for individuals with XLA has improved markedly in the last 25 years as a result of earlier diagnosis, antibiotic therapy and mainly the use of replacement gammaglobulin (intravenous or subcutaneous) allowing normal concentrations of serum IgG to be achieved [6].
Although described in 1952, the underlying genetic defect of XLA was only identified in 1994, simultaneously by Sideras et al., and Ohta et al. Bruton’s tyrosine kinase gene (BTK), a member of the Tec family of kinases, is localized on Xq21.3–Xq22 and was found to be mutated in the majority of male patients presenting with agammaglobulinemia [7, 8]. More than 85 % of patients with defect in early B cell development have X-linked agammaglobulinemia caused by mutations in BTK gene. At present, 2152 different BTK gene mutations, have been reported in the international mutation database designated BTKbase (http://structure.bmc.lu.se/idbase/BTKbase/index.php? content = index/IDbases) Half of the remaining patients, which are generally assigned to the autosomal form of the disease, have mutations in genes encoding for pre-BCR and/or BCR complex; such as IGHM, CD79a, CD79b and IGLL1. In addition, mutations in BLNK gene encoding a scaffold protein that binds to BTK or mutations in PIK3R1 gene, a kinase involved in signal transduction in multiple cell types may underly AR agammaglobulinemia. Recently, autosomal-dominant agammaglobulinemia mode of inheritance due to a recurrent mutation in TCF3, a transcription factor required for control of B cell development has been identified [9].
Barbouche et al. have reported that unlike European and North American patients, the high rate of consanguinity in North African populations is often associated to a higher prevalence of autosomal recessive forms of inheritance as compared to X-linked forms in primary immunodeficiencies [10]. This is the case for CGD and hyper IgM syndrome [11, 12]. In this context, we herein aimed to assess the frequency of XLA in North African male patients and determine the spectrum of BTK mutations.
Material and Methods
Patients
A total of 63 North African patients (50 males and 13 females) were shown to have agammaglobulinemia with less than 2 % circulating B cells. The Fifty male patients belonging to 45 unrelated families were included in this study, 50 (19 from Algeria, 16 from Morocco, and 15 from Tunisia). Ethical approval has been obtained from respective Institutional Review Boards. The referring physicians carried out structured interviews to collect demographic and clinical data.
XLA was considered according to the diagnostic criteria for XLA developed by the Joint European Society for Immunodeficiencies/Pan American Group for Immunodeficiencies Committee [13]. Male patients were considered with XLA if they had recurrent bacterial infections in the first 2 years of life, serum IgG/A/M of 2 SD below normal range for age, CD19 + B cells of <2 %. Definitive diagnosis of XLA was made if a BTK mutation was identified.
Molecular Analysis of BTK Gene
Genomic DNA was purified by phenol-chloroform from peripheral blood. Polymerase chain reaction (PCR) amplification was then carried using primers for coding exons and flanking intron sequences of the XLA gene: Eight separate polymerase chain reaction (PCR), including five regular PCR (product size < 1 kb) and three long PCR (product size of 1.8–3.9 kb), were performed using primers and conditions as previously described [14, 15]. AmpliTaq Gold DNA Polymerase System (Applied Biosystem, Foster City, CA, USA) was used for regular PCR and Expand High Fidelity PCR System (Roche Diagnostics GmbH, Mannheim, Germany) was used for long PCR. PCR products were directly sequenced using either the original PCR primers or the nested intron-flanking primers. Sequence analysis with the BTK reference sequence (Accession: U78027) were performed using the National Center for Biotechnology Information program Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/BLAST/).
In silico, prediction of missense mutations was realized by two software: SIFT (Sorting Intolerant From Tolerant) and PolyPhen-2 (Polymorphism Phenotyping).
Statistical Analysis
Kruskal–Wallis test was used to compare quantitative data. P-values less than 0.05 were considered as statistically significant. All statistical analyses were performed using STATA software, version 11.0.
Genotype-Phenotype Correlation
We classified the mutations as severe and less severe as previously described [16, 17]. Less severe mutations are missense mutations affecting non conserved residues and mutations in non-invariant splicing positions. As for disease severity, it was indicated by the age of onset and the occurrence of bloodstream infections, central nervous system infections and deep-seated infections [17].
Results
Demographic and Clinical Features
We included 50 male patients from 45 unrelated families. A BTK mutation was found in 40 patients. For the XLA patients, mean age at diagnosis was 45.42 (4–180) months, with a median age at diagnosis at 36 (18.75–60) months. Mean age at onset was 13.28 (2–48) months, with a median age at onset of 9 (6–12) months. Mean delay in diagnosis was 28.94 (0–132) months, with a median delay of 19.5 (6–43.5) months. Sixteen patients had a positive family history (death at a young age, similar clinical features or identified agammaglobulinemia in a brother or relatives from maternal side). The mean age at diagnosis among these patients was 36.31 months which was lower than that in sporadic cases (47.31 months). Clinical features were available for all XLA patients. Recurrent respiratory tract infections were the most common clinical features (37 = 92.5 %), including 22 cases of pneumonia and 6 complications by bronchiectasis. These manifestations are followed by otitis media (19 = 47.5 %), chronic diarrhea (17 = 42.50 %), arthritis (15 = 37.5 %) and meningitis (11 = 27.5 %). Five cases of septicemia and 4 cases of osteomyelitis were also reported (Tables 1 and 2).
Genetic Analysis
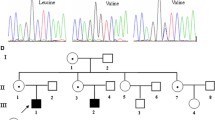
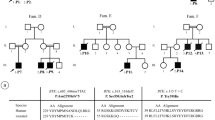
Analysis of exons 1 to 19 and the flanking intronic regions of the BTK gene revealed the presence of 33 different mutations in 40 patients from 35 unrelated families, including 12 missense mutations, 6 nonsense mutations, 6 splice-site mutations, 4 frameshift, 2 large deletions, one in frame deletion and two complex mutation (one indel and one double mutation). Among them, 17 are novel mutations including 5 missense mutations (c.308 A > C, c.1076 T > G c.1563C > G, c.1762 T > G and c.1939C > T), 3 frameshift mutations (c.1031_1037delATTACCT, c.1656-1657insCA and c.1845_1846insGT), 3 Splice site mutations (c.241-1G > A, c.975 + 1G > T, and c.1750 + 2 T > C), one nonsense mutation (c.1181C > G), one in frame deletion (c.1226_1243 delGGACTGGACAATTTGGGG), 2 complex mutations. (c.487_491del ATGGG ins T and c.1401_1402 GC > TT) and 2 large deletions (Exon 1 del and Ex2-19 del,) (Supplementary Data, Table 1 and Fig. 1).
Seventeen out of 33 mutations identified in the BTK gene were located in the Tyrosine kinase domain (TK), 6 in the Pleckstrin homology domain (PH),4 in the Src homology 2 domain (SH2) 2 in Src homology 3 domain (SH3) and 2 in Tec homology domain (TH) (Supplementary Data, Fig. 1).
In addition, 1 frameshift mutation is predicted to affect both domains SH2 and TK domains and two mutations (a large deletion and exon 1 deletion) are predicted to affect the translation of the protein.
Genotype–Phenotype Correlation
Thirty eight patients among the 40 with genetic confirmation of XLA had “severe” mutations and two had “less severe” mutations (Supplementary Data, Table 1). Genotype–phenotype correlation analysis did not reveal a clear-cut correlation between severity of mutation, as defined in Material and Methods section, and clinical phenotype. Nevertheless, a significant correlation between age at onset and type of mutations was observed. Indeed, nonsense and frame shift mutations were seemingly more severe with an early onset while aberrant splicing mutations wereless severe with delayed onset (p = 0.0067) (Table 3).
Discussion
In this work, we reviewed the clinical and genetics characteristics of a large cohort of patients diagnosed with XLA from three North African countries (Algeria, Morocco, and Tunisia). As expected a higher percentage of autosomal recessive forms in agammaglobulinemia North African patients due to the high rate of consanguinity was observed. Indeed, parental consanguinity was reported in 32.1 % of all 63 patients presenting agammaglobulinemia with less than 2 % circulating B cells, and only 17.1 % in XLA patients, similarly to consanguinity rate recorded in the corresponding general population (15–25 %) [10]. Among the 50 investigated male patients, which achieved the clinical diagnosis criteria for XLA, 40 of them have confirmed XLA diagnosis by the presence of mutations in BTK gene. The remaining 10 male patients as well as 13 females were assigned to the autosomal recessive forms of the disease (unpublished data). Thus, in our North African setting XLA represents only 63.5 %, while in Western countries BTK mutations are found in 85 % of all congenital agammaglobulinemia patients [16]. This finding confirms the hypothesis of the emergence of autosomal recessive forms in highly consanguineous North African populations.
Almost all patients developed symptoms within the first year of life. The median age at onset of disease was 9 months; this is in accordance with that observed in Iranians patients (10 months) [18]. However, the median of diagnostic age in our patients was 36 months which is less than in others studies: 42 months in Argentina, 48 months in Iran and 84 months in China [17–19].
All patients described in this study developed symptoms attributed to agammaglobulinemia including recurrent bacterial infections and severe hypogammaglobulinemia. The major clinical manifestations were an increased susceptibility to respiratory tract infections. A relatively high incidence of arthritis and diarrhea and low incidence of skin infection was also observed. When compared to other series, our cohort is clinically similar to the Iranian cohort [18] except for ENT infections and conjunctivitis that were more common in Iranian patients. We also noted a higher incidence of osteomyelitis, compared to Chinese and US cohort [5, 17], as well as a relatively high proportion of bronchiectasis compared to the Chinese cohort [17]. These features, considered as severe complications, can be explained by a delayed diagnosis in this group of patients (Mean age at diagnosis of 55 months for bronchiestasis and 32 months for osteomyelitis, vs 24 months in whole series). Other factors can intervene, such as poor patient management and lack of compliance with treatment in our region.
Genetic investigation revealed 33 different mutations in the BTK gene, among which 17 (48.48 %) were not previously reported at the best of our knowledge. Sixteen (48.48 %) out of 33 mutations were located in the Tyrosine kinase domain (TK), 6 (18.18 %) in the Pleckstrin homology domain (PH), 4 (12.12 %) in the Src homology 2 domain (SH2), 2 (6.06 %) in Src homology 3 domain (SH3), and 2 (6.06 %) in Tec homology domain (TH). These findings were in accordance with the distribution of mutations in different domains in BTKbase (http://bioinf.uta.fi/BTKbase), in which 47.1 % (590/1252) were found in the TK domain and 21.5 % (270/1252) in PH domain.
All missense mutations were predicted to be possibly or probably damaging by Polyphen2 and/or SIFT algorithms (Supplementary Data, Table 1). The previously unreported missense mutation p.Q103P, in PH domain, identified in patient P16 was predicted would be damaging by SIFT AND Polyphen 2, clinically this patient presents early age at onset (9 months) and has developed broncho-pneumopathy and diarrhea.
The arginine to cysteine substitution at codon 28 (p. R28C) of the CpG site in BTK gene was found in a Moroccan patient (p18). Arginine 28 is involved in binding potential ligands such as phospholipids [20], other mutations in this codon are cited in several studies [21–26]. This mutation showed a mild clinical phenotype in literature [27]. However, our patient presented a severe clinical phenotype, with onset at 12 months and clinical features dominated by bronchopneumopathy, diarrhea, osteomyelitis and hepatomegaly.
The Tsukada study in 2001 showed that the SH2 domain is essential for phosphorylation and activation of the phospholipase C-c and that BLNK and SH2 interaction promote this activation. This study also confirmed that mutations in SH2 domain induced XLA [28]. In our study, unreported missense mutations was identified in this domain, in an Algerian patient (p9), p.I359S, POLYPHEN 2 predicted that mutation would be damaging, this in favor of the hypothesis of a disease-causing effect. In the same domain, one novel mutation c.975 + 1G > T was described in patient P30, according to Human Splicing Finder, this mutation caused a splicing error by the change in the splice junction from GGGT to GGTT.
Two other novel mutations was found in SH1 domain: p. D521E in one Algerian patient (p8) and p.W588G in two Tunisians brothers (p12a and p12b). The D521 residue cannot be substituted by any other amino acid and mutations in this site affect the putative catalytic site in BTK [29, 30]. The W588 residue is conserved in the family of Tyrosine kinases, this suggests an essential role of these residues in the structure and function of the BTK protein [31]. Another mutation in this codon p.W588R was defined in a patient from central Europe [31]. in this domain, too, Another new missense mutation L647F was found in a Moroccan patient (p17), who presented at the time of diagnosis a pancytopenia and some infections such as pneumonia, otitis and Diarrhea. Also in this domain, the unreported in frame deletion (c.1226_1243delGGACTGGACAATTTGGGG) lead to excision of 5 amino acids which are conserved (Supplementary Data, Fig. 2), and was found in two Algerian brothers (p06a and p06b). Clinically the patient 06a had a severe phenotype characterized by the early onset of the disease at the age of 9 months and meningitis infection; his brother presented at 8 months with pneumonia, a trisomy 21 and cardiomyopathy. Interestingly, in the SH1 domain, the same mutation c.1631 + 1G > A had been identified in two Tunisians brothers and a one Moroccan patient. This mutation has been previously reported [19].
We expected a correlation between the severity of mutations (based on the type of mutations) and the clinical phenotype, as it was shown in several studies [17, 23, 32, 33]. Statistical analysis showed no significant correlation between severity of mutations and clinical phenotype. In contrast, significant correlations were found between type of mutation and age at onset and at diagnosis.
Conclusion
In summary, this is the largest North African cohort describing mutations in the BTK gene. The characteristics of BTK mutations were generally maintained to that reported in other regions and several novel mutations are reported. Molecular analysis of the BTK gene is an enabling tool for the diagnosis of XLA and is essential for genetic counseling. This study also highlighted the emergence of autosomal recessive forms in the North African region.
References
Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9:722–8.
Vihinen M, Kwan SP, Lester T, Ochs HD, Resnick I, Väliaho J, et al. Mutations of the human BTK gene coding for bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat. 1999;13:280–5.
Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood. 2000;96:610–7.
Chapel H, Geha R, Rosen F. IUIS PID (Primary Immunodeficiencies) classification committee. Primary immunodeficiency diseases: an update. Clin Exp Immunol. 2003;132:9–15.
Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85:193–202.
Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr. 2002;141:566–71.
Sideras P, Müller S, Shiels H, Jin H, Khan WN, Nilsson L, et al. Genomic organization of mouse and human Bruton’s agammaglobulinemia tyrosine kinase (Btk) loci. J Immunol. 1994;153:5607–17. Baltim. Md 1950.
Ohta Y, Haire RN, Litman RT, Fu SM, Nelson RP, Kratz J, et al. Genomic organization and structure of Bruton agammaglobulinemia tyrosine kinase: localization of mutations associated with varied clinical presentations and course in X chromosome-linked agammaglobulinemia. Proc Natl Acad Sci U S A. 1994;91:9062–6.
Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
Barbouche M-R, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. 2011;1238:42–52.
El Kares R, Barbouche MR, Elloumi-Zghal H, Bejaoui M, Chemli J, Mellouli F, et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J Hum Genet. 2006;51:887–95.
Ouadani H, Ben-Mustapha I, Ben-Ali M, Ben-Khemis L, Larguèche B, Boussoffara R, et al. Novel and recurrent AID mutations underlie prevalent autosomal recessive form of HIGM in consanguineous patients. Immunogenetics. 2015;68(1):19–28.
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190–7. Orlando Fla.
Yip KL, Chan SY, Ip WK, Lau YL. Bruton’s tyrosine kinase mutations in 8 Chinese families with X-linked agammaglobulinemia. Hum Mutat. 2000;15:385.
Chan K-W, Chen T, Jiang L, Fok SF-S, Lee T-L, Lee B-W, et al. Identification of Bruton tyrosine kinase mutations in 12 Chinese patients with X-linked agammaglobulinaemia by long PCR-direct sequencing. Int J Immunogenet. 2006;33:205–9.
Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005;203:216–34.
Lee PPW, Chen T-X, Jiang L-P, Chan K-W, Yang W, Lee B-W, et al. Clinical characteristics and genotype-phenotype correlation in 62 patients with X-linked agammaglobulinemia. J Clin Immunol. 2010;30:121–31.
Aghamohammadi A, Fiorini M, Moin M, Parvaneh N, Teimourian S, Yeganeh M, et al. Clinical, immunological and molecular characteristics of 37 Iranian patients with X-linked agammaglobulinemia. Int Arch Allergy Immunol. 2006;141:408–14.
Basile N, Danielian S, Oleastro M, Rosenzweig S, Prieto E, Rossi J, et al. Clinical and molecular analysis of 49 patients with X-linked agammaglobulinemia from a single center in Argentina. J Clin Immunol. 2009;29:123–9.
Fukuda M, Kojima T, Kabayama H, Mikoshiba K. Mutation of the pleckstrin homology domain of Bruton’s tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5-tetrakisphosphate binding capacity. J Biol Chem. 1996;271:30303–6.
Hashimoto S, Tsukada S, Matsushita M, Miyawaki T, Niida Y, Yachie A, et al. Identification of Bruton’s tyrosine kinase (Btk) gene mutations and characterization of the derived proteins in 35 X-linked agammaglobulinemia families: a nationwide study of Btk deficiency in Japan. Blood. 1996;88:561–73.
Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, et al. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol. 1998;161:3925–9. Baltim. Md 1950.
Holinski-Feder E, Weiss M, Brandau O, Jedele KB, Nore B, Bäckesjö CM, et al. Mutation screening of the BTK gene in 56 families with X-linked agammaglobulinemia (XLA): 47 unique mutations without correlation to clinical course. Pediatrics. 1998;101:276–84.
Wang Y, Kanegane H, Sanal O, Ersoy F, Tezcan I, Futatani T, et al. Bruton tyrosine kinase gene mutations in Turkish patients with presumed X-linked agammaglobulinemia. Hum Mutat. 2001;18:356.
Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton’s tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114:141–9.
Tani SM, Wang Y, Kanegane H, Futatani T, Pinto J, Vilela MM, et al. Identification of mutations of Bruton’s tyrosine kinase gene (BTK) in Brazilian patients with X-linked agammaglobulinemia. Hum Mutat. 2002;20:235–6.
Conley ME, Mathias D, Treadaway J, Minegishi Y, Rohrer J. Mutations in btk in patients with presumed X-linked agammaglobulinemia. Am J Hum Genet. 1998;62:1034–43.
Kanegane H, Futatani T, Wang Y, Nomura K, Shinozaki K, Matsukura H, et al. Clinical and mutational characteristics of X-linked agammaglobulinemia and its carrier identified by flow cytometric assessment combined with genetic analysis. J Allergy Clin Immunol. 2001;108:1012–20.
Mattsson PT, Vihinen M, Smith CI. X-linked agammaglobulinemia (XLA): a genetic tyrosine kinase (Btk) disease. BioEssays. 1996;18:825–34 News Rev. Mol. Cell. Dev. Biol.
Vorechovský I, Vihinen M, de Saint BG, Honsová S, Hammarström L, Müller S, et al. DNA-based mutation analysis of Bruton’s tyrosine kinase gene in patients with X-linked agammaglobulinaemia. Hum Mol Genet. 1995;4:51–8.
Kristufek D, Aspalter RM, Eibl MM, Wolf HM. Characterization of novel Bruton’s tyrosine kinase gene mutations in Central European patients with agammaglobulinemia. Mol Immunol. 2007;44:1639–43.
Teimourian S, Nasseri S, Pouladi N, Yeganeh M, Aghamohammadi A. Genotype-phenotype correlation in Bruton’s tyrosine kinase deficiency. J Pediatr Hematol Oncol. 2008;30:679–83.
López-Granados E, de Pérez Diego R, Ferreira Cerdán A, Fontán Casariego G, García Rodríguez MC. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol. 2005;116:690–7.
Acknowledgments
We would like to thank the patients and their families for their collaboration. This work was supported by ACIP: A-07-2011 (Actions Concertées Inter Pasteuriennes) from the International Network of Pasteur Institutes (RIIP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Zahra Aadam and Nadia Kechout contributed equally to this work.
Rights and permissions
About this article

Cite this article
Aadam, Z., Kechout, N., Barakat, A. et al. X-Linked Agammagobulinemia in a Large Series of North African Patients: Frequency, Clinical Features and Novel BTK Mutations. J Clin Immunol 36, 187–194 (2016). https://doi.org/10.1007/s10875-016-0251-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-016-0251-z