
Abstract
High-frequency measurements of dibromomethane (CH2Br2) and bromoform (CHBr3) at Hateruma Island, in the subtropical East China Sea, were performed using automated preconcentration gas chromatography/mass spectrometry. Their baseline concentrations, found in air masses from the Pacific Ocean, were 0.65 and 0.26 ppt, respectively, in summer and 1.08 and 0.87 ppt, respectively, in winter. Air masses transported from Southeast Asia were rich in bromocarbons, suggesting strong emissions in this area. The passage of cold fronts from the Asian continent was associated with sharp increases in observed concentrations of bromocarbons derived from coastal regions of the continent. Comparison of the relationships between [CH2Br2]/[CHBr3] and [CHBr3] in the Hateruma Island data with those in monthly mean data from 14 globally distributed U.S. National Oceanic and Atmospheric Administration ground stations suggested that these gases are produced primarily from a common process on a global scale.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Bromoform (CHBr3) and dibromomethane (CH2Br2), which undergo photolytic degradation and react with OH to produce inorganic bromine, are the large contributors of organic bromine from the ocean to the atmosphere, where it can affect stratospheric and tropospheric ozone chemistry (Carpenter and Liss 2000; Montzka and Reimann 2011). These naturally produced ozone-depleting substances (ODS) are attracting more interest as concentrations of anthropogenic ODS decrease under the provisions of the Montreal Protocol.
The major sources of these bromocarbons are believed to be seaweed or macroalgae, followed by phytoplankton and other biological sources (Manley et al. 1992; Sturges et al. 1992; Tokarczyk and Moore 1994; Moore et al. 1996; Carpenter and Liss 2000; Laturnus 2001); their sea–air fluxes decrease geographically in the order coastal > upwelling waters > shelf > open ocean (Carpenter et al. 2009). Many uncertainties remain, however, with regard to their production mechanisms and the factors affecting their emissions.
Reported atmospheric concentrations are mostly in the range of 0.5–2 ppt for CH2Br2 and 0.2–10 ppt for CHBr3, with higher concentrations in coastal areas (Atlas et al. 1992, 1993; Schauffler et al. 1999; Carpenter et al. 2003; Yokouchi et al. 2005; O’Brien et al. 2009; Hossaini et al. 2013). In many observations, concentrations of these gases show fairly good correlations, suggesting shared sources. Owing to the shorter lifetime of CHBr3 (~24 days) compared to that of CH2Br2 (~123 days) (Montzka and Reimann 2011), background levels of CHBr3 are generally lower than those of CH2Br2, and CHBr3 shows a seasonal variation with larger relative amplitude.
Current estimates of their global emissions vary considerably; estimates for CH2Br2 range from 57 to 280 Gg Br/y, and those for CHBr3 range from 120 to 1400 Gg Br/y (Quack and Wallace 2003; Yokouchi et al. 2005; Warwick et al. 2006; Butler et al. 2007; O’Brien et al. 2009; Liang et al. 2010; Montzka and Reimann 2011; Ordonez et al. 2012; Ziska et al. 2013); as a result, it is difficult to assess the impact of these bromocarbons on stratospheric ozone depletion and possible biogenic feedback under climate change. Some studies have used sea-to-air fluxes based on bottom-up inventories, whereas others have used fluxes based on top-down inventories, obtained by using airborne measurements or emission ratios inferred from atmospheric observations. The uncertainties in these estimates derive from difficulties caused by their highly heterogeneous emissions. Thus, to understand the overall situation of these compounds in the atmosphere, it is important to collect data with high spatial and temporal resolution and to make use of longer-term observations representative of broad regions of the global atmosphere.
Here we report high-frequency long-term measurements of CH2Br2 and CHBr3 obtained on an island that is occasionally affected by air masses from the Asian continental coast, the Pacific Ocean, tropical Southeast Asia, and local coral reefs. We also discuss the global dynamics of these bromocarbons by comparing the island data with long-term observations carried out at U.S. National Oceanic and Atmospheric Administration (NOAA) ground-based stations at multiple sites across the globe.
2 Observations
High-frequency measurements of CH2Br2 and CHBr3 in the atmosphere were done at a remote monitoring station of the National Institute for Environmental Studies (NIES) on Hateruma Island (24°4′N, 123°49′E, about 10 m a.s.l.) (http://db.cger.nies.go.jp/gem/en/warm/Ground/st01.html). Hateruma Island is a small subtropical island in the pathway of the warm Kuroshio Current in the East China Sea. The island is surrounded by fringing coral reefs with poor seaweed growth.
Air samples were mostly taken from a 39-m tower (air intake height, 46.5 m a.s.l.), and sometimes from a 10-m tower (17.5 m a.s.l.) for comparison. The samples were analyzed by an automated preconcentration gas chromatography–mass spectrometry (GC–MS) instrument (Agilent 6890/5973) designed for hourly measurements of natural and anthropogenic halocarbons. Details of the sampling and analytical methods have been published elsewhere (Enomoto et al. 2005; Yokouchi et al. 2006, 2011). Selected ion monitoring was employed, and m/z (mass to charge) ratios of 174 and 173 were monitored for quantification of CH2Br2 and CHBr3 respectively. After every five air analyses, a gravimetrically prepared standard gas (Taiyo Nippon Sanso Co. Ltd.) containing 100 ppt each of CH2Br2 and CHBr3 was analyzed for quantification. Sometimes concentrations of low-concentration standard gases decrease in standard gas cylinders, so that subsequent calibration by comparison with standards prepared later is important. For the target period of this study (December 2007 to November 2008), such comparisons confirmed no changes in the CH2Br2 and CHBr3 concentrations of the standards used at Hateruma. To avoid any possible carry-over or memory effect, samples analyzed immediately after the standard gas were excluded from the following analysis. Thus, data from 16 samples each sampling day are discussed here.
3 Results and discussion
3.1 Seasonal variation and source apportionment of CH2Br2 and CHBr3 observed at Hateruma Island
The CH2Br2 and CHBr3 concentration datasets measured at Hateruma Island from December 2007 to November 2008 are shown by season in Figs. 1 and 2, respectively. For comparison, HCFC-22 concentration data are also included in Fig. 1, because this compound is an anthropogenic fluorocarbon that can serve as an indicator of the influence of air masses from the Asian continent (Yokouchi et al. 2006). Both bromocarbons showed clear seasonal variation as well as occasional abrupt short-term increases. The average concentration of CH2Br2 was 1.20 ppt (1σ, 0.10 ppt) in winter (December–February) and 1.06 ppt (1σ, 0.22 ppt) in summer (June–August), and that of CHBr3 was 1.28 ppt (1 σ, 0.30 ppt) in winter and 0.91 ppt (1 σ, 0.55 ppt) in summer. Similar seasonal variations, lower in summer than in winter, and higher variability of CHBr3, observed at various sites, have been explained previously by seasonal changes in atmospheric reactive losses and the shorter lifetime of CHBr3 (Yokouchi et al. 1996, 2005; Carpenter and Liss 2000; Brinckmann et al. 2012).

Hourly variations of CH2Br2 measured at Hateruma (circles) and of HFC-22 (crosses). From the top, December 2007 to February 2008, March to May 2008, June to August 2008, September to November 2008. The air samples were collected from a 39-m (gray symbols) or 10-m tower (black symbols). A–L are explained in the text

Hourly variation of CHBr3 measured at Hateruma. Other information is the same as in Fig. 1
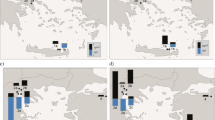
Compared to NOAA monthly mean data from Cape Kumakahi, Hawaii, which is at a similar latitude (19.5°N) and has a comparable sampling altitude to Hateruma Island (Hossaini et al. 2013), the winter and summer averages of CH2Br2 and CHBr3 at Hateruma are a little higher; at Cape Kumakahi, the monthly mean CH2Br2 concentration is 0.95–1.05 ppt in winter and 0.65–1.05 ppt in summer, and that of CHBr3 is 1.17–1.26 ppt in winter and 0.80–0.87 ppt in summer. These differences between the sites might be due to the occasional short-term enhancement (peaks lasting hours to days) observed at Hateruma, which are considered to reflect particular emission sources affecting the site. Because macroalgae are scarce in the vicinity of Hateruma Island, these short-term bromocarbon increases may be caused by long-range transport from strong source regions or by a local source other than macroalgae. To identify such possible source regions, we used “footprint indicator” results, shown in Fig. 3, for selected air masses (A–L in Figs. 1 and 2) We took the mean indicator of 10,000 particles in one degree grid cells and within 250 m above the surface as the footprint indicator. Such a layer was treated as foot print layer (e,g, Cooper et al. 2010). The particles were released 100 m above the surface of Hateruma station and their trajectories were calculated back in time for 10 days. The Lagrangian particle dispersion model is based on FLEXPART (https://flexpart.eu/). The wind fields of the Global Data Assimilation System (https://ready.arl.noaa.gov/gdas1.php) were used with the model.

In winter and spring, we observed many CH2Br2 and CHBr3 peaks characterized by a sharp initial rise, most of which were accompanied by HCFC-22 peaks. In these seasons, the outflow of pollutants from the Asian continent to Japan, associated with the passage of cold fronts moving from the northwest to southward, cause occasional increases of HCFC-22 and other pollutants at Hateruma (Yokouchi et al. 2006). The air masses corresponding to the observed peaks (e.g., B, C in Fig. 1) passed over coastal regions of the Yellow Sea or the East China Sea before arriving at Hateruma (Fig. 3b, c). Thus, bromocarbons emitted from coastal waters of the Asian continent, which are rich in macroalgae, were also swept up by the cold fronts, resulting in enhanced bromocarbon concentrations coupled with increases in HCFC-22 emitted from coastal megacities. However, some short-term increases of bromocarbons were also observed with little concurrent enhancement of HCFC-22 (e.g., D in Fig. 1). These air masses had traveled over coastal regions of the Sea of Japan, where there are no large cities (see Fig. 3d). In contrast, baseline concentrations were observed when the air mass came from the Pacific Ocean east and south of Hateruma Island (e.g., A, CH2Br2, 1.08 ppt and CHBr3, 0.87 ppt; and E, CH2Br2, 0.88 ppt and CHBr3, 0.61 ppt).
In summer, we observed broad bromocarbon peaks with amplitudes of ~0.5 ppt for CH2Br2 and ~1 ppt for CHBr3, with no enhancement of HCFC-22 (e.g., H, I in Fig. 1). Hateruma Island is rarely affected by polluted air masses from the Asian continent in summer, so the observed HCFC-22 concentrations are low. The footprint indicator (or back trajectory) showed that these bromocarbon-rich air masses were transported from tropical Southeast Asia (Fig. 3h, i). This area has attracted interest as a possible hotspot for the transport of bromocarbons to the upper troposphere or stratosphere by strong convection (Tegtmeier et al. 2012), although some recent studies have reported that CH2Br2 and CHBr3 concentrations observed in Southeast Asia are not particularly high except in coastal regions (Hossaini et al. 2013; Nadzir et al. 2014). However, the remarkable enhancement of bromocarbons in air masses transported long distances from Southeast Asia indicates that the widespread bromocarbon sources in Southeast Asia might make a large contribution as a whole. This is a likely scenario, considering that Southeast Asia is a warm region and comprises more than ten thousand islands; thus, the coastal zone is large due to the long coastlines. Air masses from the Pacific Ocean (e.g., G) gave summer baseline concentrations of 0.65 ppt for CH2Br2 and 0.26 ppt for CHBr3.
In autumn, the wind systems affecting Hateruma are variable. Footprint indicators showed that the enhancement of CH2Br2 and CHBr3 was mostly attributable to long-range transport from Southeast Asia (e.g., J) or from coastal areas of the Sea of Japan (e.g., L). Baseline concentrations were observed when the air masses came from the Pacific Ocean (e.g., K, CH2Br2, 0.75 ppt, and CHBr3, 0.47 ppt).
Comparison of alternative measurements between the 39-m and 10-m towers showed that a considerable difference in bromocarbon concentrations existed between them only in data from late May to early June. For example, the average CH2Br2 and CHBr3 concentrations from the 39-m tower during 17:00–21:00 Japan Standard Time (JST) on 25 May were 0.77 ppt and 0.52 ppt, respectively, and those from the 10-m tower were 1.37 ppt and 3.23 ppt, respectively (F in Fig. 1). Large differences between the towers were observed when a weak wind was blowing from the east, suggesting that the coral reefs extending eastward from Hateruma Island were a possible source of the enhanced bromocarbon concentrations measured from the 10-m tower. The corresponding footprint (F in Fig. 3) showed that the air mass was transported from the Pacific Ocean, suggesting little influence on the bromocarbons concentrations by long-range transport from other source regions.
3.2 Relationships between the CH2Br2 and CHBr3 concentrations observed at Hateruma
The faster degradation of CHBr3 relative to CH2Br2 during transport causes the [CH2Br2]/[CHBr3] ratio to increase with transport time from the sources. Mixing with background air also increases this ratio, because the chemically aged background air has a relatively lower concentration of the shorter lived CHBr3.
If dilution or mixing is negligible and chemical decay is dominant, then the concentrations of two compounds, [A] and [B], at time t can be defined as follows:
where [A]0 and [B]0 represent their initial concentrations, kA and kB are the pseudo-first-order rate constants, or lifetime reciprocals, and t is the time since the injection of the compounds into the sampled air mass.
By combining Eqs. (1) and (2), t can be eliminated, yielding the following equation:
Thus, the natural logarithm of the ratio [B]/[A] is linearly related to the natural logarithm of [A], and the slope of the resulting line is determined by the rate constants, or the reciprocals of the lifetime (τ). By using the lifetimes of CH2Br2 and CHBr3 (123 days and 24 days, respectively), the slope can be calculated to be −0.80. Here, we refer to the line defined by Eq. (3) as the “chemical decay line”.
In the annual dataset from Hateruma, ln([CH2Br2]/[CHBr3]) and ln([CHBr3]) are strongly correlated (Fig. 4a), the slope of the regression line is −0.66, and the coefficient of determination (R2) is 0.93. (Note that two variables (A and B) with random Gaussian distributions also show good correlation between ln(A) and ln(B/A) but with a slope of −1, given a mean and standard deviation identical to the Hateruma data.) Although this result might suggest that chemical decay dominates the relationship, the regression slope (−0.63) is not consistent with the chemical decay line defined by Eq. 3 (m = −0.80). The chemical decay line from the globally dominant source(s) is expected to pass through the background (or most aged) airmass, and to have a slope of −0.80. The broken line in Fig. 4a meets these conditions. This chemical decay line matches the lower-left margin of all data obtained at Hateruma, although many of the data are deviated above and to the right. A simple simulation has shown that mixing (or dilution) of the airmass on this chemical decay line would cause a slight deflection to the left, not the right, of this line. Therefore, those data appearing above and to the right of this decay line suggest higher CHBr3 to CH2Br2 ratios in sources affecting Hateruma than in sources dominating this ratio in the background airmass.

Relationship between [CH2Br2]/[CHBr3] and [CHBr3] in individual samples collected at Hateruma. (a) Full year (these data appear in panels (b) – (e) as gray points), (b) December–February (black), (c) March–May (green), (d) June–August (pink), (e) September–November (blue). The solid line in (a) is the regression line for the complete dataset. Broken lines show possible chemical decay lines, with a slope of −0.80, for each season. Points A–L are explained in the text
To identify seasonal features, we compared the ln([CH2Br2]/[CHBr3])–ln([CHBr3]) relationship in the data of each season with the annual dataset (Fig. 4b–e). In all seasons, we found good correlations between [CH2Br2]/[CHBr3] and [CHBr3], with regression slopes similar to that for the whole annual dataset (Fig. 4a): winter, −0.72 (R2 = 0.91); spring, −0.71 (R2 = 0.94); summer, −0.70 (R2 = 0.96); and autumn, −0.65 (R2 = 0.94). The summer data were most scattered and included the highest [CH2Br2]/[CHBr3] ratios (and lowest CHBr3 concentrations), indicating the highest reactive losses. In contrast, the winter data were scattered within a narrower range of [CH2Br2]/[CHBr3]. The narrower scatter may be because slower oxidation rates during winter limit the photochemical age of the air and the [CH2Br2]/[CHBr3] ratio. It is noteworthy that the winter data were shifted positively (toward the upper right of the figure) compared with the data of other seasons. Possible explanations for this are discussed in the next section.
Points A–L in Fig. 4 show the relationship between [CH2Br2]/[CHBr3] and [CHBr3] in air masses A–L in Figs. 1, 2 and 3. The baseline data in each season (A, E, G, K) had the highest [CH2Br2]/[CHBr3] ratios (and lowest [CHBr3]) of the season, as expected for aged air masses not influenced recently by substantial emissions of either CH2Br2 or CHBr3. A line with slope equal to that expectd from chemical loss in each season is drawn so that it passes through these baseline data in each figure. The Asian continental air masses transported by passing cold fronts (B, C, D in winter and spring) had the lowest [CH2Br2]/[CHBr3] levels and highest [CHBr3], reflecting little chemical decay since emission because of the short transport time, while the air masses from Southeast Asia (H, I, J in summer and autumn) had moderately low [CH2Br2]/[CHBr3] ratios. Thus, coastal-affected airmasses have [CH2Br2]/[CHBr3] ratios that tend to be deviated toward higher [CHBr3] from the line defined by the [CH2Br2]/[CHBr3] ratio measured in the background atmosphere and chemical loss.
Comparison of the measurements from the 39-m and 10-m towers (F in spring) showed a remarkable difference in the relationship depending on the sampling height. [CHBr3] and [CH2Br2]/[CHBr3] observed from the 39-m tower, calculated by using the data on 25 May, were 0.52 ppt and 1.46, respectively, whereas from the 10-m tower they were 3.23 ppt and 0.43, respectively. This is consistent with an interpretation that the 10 m-tower result was more sensitive to local emissions while the 39 m-tower result indicated aged airmass.
3.3 Consideration of global sources
As described in section 3.2, the relationship between [CH2Br2]/[CHBr3] and [CHBr3] observed at Hateruma seems to reflect the globally predominant sources of the bromocarbons as well as additional coastal sources on local ~ regional scales. To examine the relationship on a global scale, we compared NOAA global observation data (14-yr monthly data of CH2Br2 and CHBr3 from 14 stations; see Table 1) with the Hateruma data in summer and winter (Fig. 5).

Relationship between [CH2Br2]/[CHBr3] and [CHBr3] in the NOAA station data and at Hateruma. (a) July monthly means for NH stations (black solid circles), January monthly means for SH stations (black open circles) and a summer baseline for Hateruma (red circle, G in Fig. 4) plotted with the whole Hateruma summer data (pink circles).; (b) January monthly means for NH stations (black solid circles), July monthly means for SH stations (black open circles) and a winter baseline for Hateruma (red circle, A in Fig. 4) plotted with the whole Hateruma winter data (gray open circles). The lines are explained in the text. Refer to Table 1 for the NOAA station abbreviations
By comparing the [CH2Br2]/[CHBr3] versus [CHBr3] relationship in NOAA monthly mean data for summer (July monthly means for Northern Hemisphere (NH) stations, January monthly mean for Southern Hemisphere (SH) stations) with summertime Hateruma data (see Fig. 4d), we can see that the NOAA data fall mostly on the chemical decay line that was drawn to fit the Hateruma summer backgroud data (slope, −0.80) (shown as broken line in Fig. 5a). The small deviations of MHD, BRW, and THD data from the line likely reflects the close proximity of these stations to algal sources of bromocarbons that have somewhat unique CH2Br2/CHBr3 ratios. It is amazing that a single chemical decay line works for characterizing the relative concentration of these two bromocarbons in aged air masses at many different remote locations all over the world. This finding strongly suggests that largest sources of CH2Br2 and CHBr3 are derived from a common process on a global scale. However, the emission strengths and emission ratios of these two bromocarbons from the ocean are known to be highly variable depending on the region; reported emission rate ratios of CH2Br2 to CHBr3 range from <0.1 to 0.43, with lower values in coastal waters [Yokouchi et al. 2005; Carpenter et al. 2009; Hughes et al. 2009; Liu et al. 2011; Ziska et al. 2013]. This apparent contradiction between “common source(s)” and “variable emission rate ratios” may be reconcilable, if the sources responsible for the majority of the CH2Br2 and CHBr3 measured in the remote atmosphere have a relationship between the CH2Br2 / CHBr3 oceanic emission rate ratio (ECH2Br2 / ECHBr3) and the CHBr3 emission rate (ECHBr3) which is fairly similar to that between [CH2Br2]/[CHBr3] and [CHBr3] of the chemical decay line. Here we consider the emitted source gases to have the same initial concentrations of bromocarbons for the sampled airmass. The former assumption may be partly supported by seawater measurements made by Carpenter and Liss (2000), who reported that the concentration ratio [CH2Br2]/[CHBr3] decreases in seawater as the concentration of CHBr3 increases.
Likewise, when the NOAA monthly data for winter (January monthly means for NH sites, July monthly means for SH sites) are plotted together with the Hateruma winter data (Fig. 5b), where the high [CHBr3] and low [CH2Br2]/[CHBr3] at ALT and BRW are reflecting no photolysis and very little OH under no sunlight at these sites during this season. Most data points show a positive shift (toward the upper right) compared with the summer data and relative to the summer chemical decay line (broken line), as in the case of the Hateruma data. This winter shift might be due to (1) a seasonal difference in relative reactivity, or (2) a seasonal difference in the emission rate ratio. With regard to the first possibility, the loss ratios in the marine boundary layer in mid-latitudes by season (14 days for CHBr3 and 80 days for CH2Br2 in summer, 86 days for CHBr3 and 620 days for CH2Br2 in winter) (Carpenter and Reimann 2015) suggests a higher reactivity ratio of CHBr3 /CH2Br2 in winter by 25 %, resulting in a steeper slope (−0.86) of the chemical decay line. With this steeper slope, we can position a chemical decay line that passes close to most of the NOAA observations (solid line in Fig. 5b). The above seasonal loss ratios in the marine boundary layer at mid-latitudes implies a slope of −0.83 for the chemical decay line in summer, which is also shown in solid line in Fig. 5a. With regard to the second possibility, if the chemical decay line estimated for the summer data is parallel shifted toward the upper right, it can be made to pass through the data points of seven of the NH sites (dot-and-dash line in Fig. 5b). However, the NH sites ALT and BRW and the four SH sites fall below and to the left of this line. Thus, the first possibility is likely to be more consistent with the summer/winter difference in the relationship of [CH2Br2]/[CHBr3] vs [CHBr3], although that should accompany seasonal change of the emission rate ratios in certain instances.
We now have a picture of the relationship between [CH2Br2]/[CHBr3] and [CHBr3] for global predominant bromocarbon sources. In summer, plots of [CH2Br2]/[CHBr3] against [CHBr3] in an aged air mass should fall on the chemical decay line defined by the following equation:
Here, the intercept was obtained by extrapolation of the broken line in Fig. 5a. In winter, the chemical decay line is a little steeper, and the plots of [CH2Br2]/[CHBr3] versus [CHBr3] show a narrower dynamic range.
The results of the present study strongly suggest that the main sources of CH2Br2 and CHBr3 to the global atmosphere are derived from a common process. Variable emission ratios of CH2Br2 /CHBr3 can be compatible with their having common sources if the emission ratio CH2Br2/CHBr3 decreases as the CHBr3 emission rate increases, thus showing a relationship similar to the chemical decay relationship in the atmosphere. On the other hand, results in some airmasses influenced by coastal processes deviated from the chemical decay line fitting the background air, suggesting the possiblity that coastal (or macroalgal) sources do not dominate the tropospheric emissions of CH2Br2 and CHBr3 throughout the globe.
4 Conclusion
The full-year high-frequency datasets of CH2Br2 and CHBr3 at Hateruma Island allowed precise analyses of their sources and dynamics in the atmosphere. By combining these data with NOAA global observation data (14-yr monthly data from 14 ground stations), we obtained new insight into the global sources of these bromocarbons and their chemical degradation.
Our major findings are as follows:
-
1.
High-frequency measurements showed that mid-latitude baseline concentrations of CH2Br2 and CHBr3 at Hateruma were 0.65 ppt and 0.26 ppt, respectively, in summer and 1.08 ppt and 0.87 ppt, respectively, in winter.
-
2.
Hateruma is influenced by multiple different regions emitting bromocarbons over the course of a year. Air masses transported from Southeast Asia in summer were enriched in the bromocarbons; the concentrations of CH2Br2 and CHBr3 were enhanced relative to the baseline by ~0.5 ppt and ~1.0 ppt, respectively, suggesting large emissions of bromocarbons from a strong convection area. The passage of cold fronts from the Asian continent was associated with a sharp increase in bromocarbons derived from coastal regions of the Asian continent. Because the cold fronts move rapidly and have an extremely reduced mixing height [e.g. Sakai et al. 2005], chemical loss and dilution are suppressed, resulting in little change in the concentrations and ratios of atmospheric components during transport.
-
3.
The relationship between [CH2Br2]/[CHBr3] and [CHBr3] at Hateruma could be explained by their chemical decay in the atmosphere with a fairly consistent CH2Br2/CHBr3 initial emission ratio, and some additional coastal effects and mixing. In winter, the plots of [CH2Br2]/[CHBr3] against [CHBr3] were shifted toward relatively higher CH2Br2.
-
4.
The multi-year monthly mean values from 14 NOAA stations in mid-summer, other than MHD and THD, fell on the chemical decay line drawn to fit the Hateruma summer data, suggesting that CH2Br2 and CHBr3 are produced primarily from a common process on a global scale. The same was true of the wintertime data, except that the slope was a little steeper, which was partly explained by a lower reactivity ratio of CH2Br2 relative to CHBr3 in winter.
-
5.
Coastal sites such as MHD and THD showed somewhat different relationship of [CH2Br2] and [CHBr3] from the chemical decay line that fits results from a number of remote sites across the globe, suggesting that results from these coastal sites do not represent the majority of emissions reaching the troposphere.
References
Atlas, E., Schauffler, S.M., Merrill, J.T., Hahn, C.J., Ridley, B., Walega, J., Greenberg, J., Heidt, L., Zimmerman, P.: Alkyl nitrate and selected halocarbon measurements at Mauna-Loa-observatory. Hawaii. J. Geophys. Res.-Atmos. 97(D10), 10331–10348 (1992)
Atlas, E., Pollock, W., Greenberg, J., Heidt, L., Thompson, A.M.: Alkyl nitrates, nonmethane hydrocarbons, and halocarbon gases over the equatorial pacific-ocean during saga-3. J. Geophys. Res.-Atmos. 98(D9), 16933–16947 (1993)
Brinckmann, S., Engel, A., Boenisch, H., Quack, B., Atlas, E.: Short-lived brominated hydrocarbons—observations in the source regions and the tropical tropopause layer. Atmos. Chem. Phys. 12(3), 1213–1228 (2012)
Butler, J.H., King, D.B., Lobert, J.M., Montzka, S.A., Yvon-Lewis, S.A., Hall, B.D., Warwick, N.J., Mondeel, D.J., Aydin, M., Elkins, J.W.: Oceanic distributions and emissions of short-lived halocarbons. Glob. Biogeochem. Cycles 21, GB1023 (2007)
Carpenter, L.J., Liss, P.S.: On temperate sources of bromoform and other reactive organic bromine gases. J. Geophys. Res.-Atmos. 105(D16), 20539–20547 (2000)
Carpenter, L.J., Reimann, S.: Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, Chapter 1 in Scientific Assessment of Ozone Depletion: 2014. Global Ozone Research and Monitoring Project – Report no. 55. World Meteorological Organization, Geneva (2015)
Carpenter, L.J., Liss, P.S., Penkett, S.A.: Marine organohalogens in the atmosphere over the Atlantic and Southern Oceans. J. Geophys. Res.-Atmos. 108(D9), 4256 (2003)
Carpenter, L.J., Jones, C.E., Dunk, R.M., Hornsby, K.E., Woeltjen, J.: Air-sea fluxes of biogenic bromine from the tropical and North Atlantic Ocean. Atmos. Chem. Phys. 9(5), 1805–1816 (2009)
Cooper, O.R., Parrish, D.D., Stohl, A., Trainer, M., Nedelec, P., Thouret, V., Cammas, J.P., Oltmans, S.J., Johnson, B.J., Tarasick, D., Leblanc, T., McDermid, I.S., Jaffe, D., Gao, R., Stith, J., Ryerson, T., Aikin, K., Campos, T., Weinheimer, A., Avery, M.A.: Increasing springtime ozone mixing ratios in the free troposphere over western North America. Nature 463, 344–348 (2010)
Enomoto, T., Yokouchi, Y., Izumi, K., Inagaki, T.: Development of an analytical method for atmospheric halocarbons and its application to airborne observation. J. Jpn. Soc. Atmos. Environ. 40, 1–8 (2005)
Hossaini, R., Mantle, H., Chipperfield, M.P., Montzka, S.A., Hamer, P., Ziska, E., Quack, B., Krueger, K., Tegtmeier, S., Atlas, E., Sala, S., Engel, A., Boenisch, H., Keber, T., Oram, D., Mills, G., Ordonez, C., Saiz-Lopez, A., Warwick, N., Liang, Q., Feng, W., Moore, E., Miller, B.R., Marecal, V., Richards, N.A.D., Dorf, M., Pfeilsticker, K.: Evaluating global emission inventories of biogenic bromocarbons. Atmos. Chem. Phys. 13(23), 11819–11838 (2013)
Hughes, C., Chuck, A.L., Rossetti, H., Mann, P.J., Turner, S.M., Clarke, A., Chance, R., Liss, P.S.: Seasonal cycle of seawater bromoform and dibromomethane concentrations in a coastal bay on the western Antarctic Peninsula. Glob. Biogeochem. Cycles 23, GB2024 (2009)
Laturnus, F.: Marine macroalgae in polar regions as natural sources for volatile organohalogens. Environ. Sci. Pollut. Res. 8(2), 103–108 (2001)
Liang, Q., Stolarski, R.S., Kawa, S.R., Nielsen, J.E., Douglass, A.R., Rodriguez, J.M., Blake, D.R., Atlas, E.L., Ott, L.E.: Finding the missing stratospheric Br-y: a global modeling study of CHBr3 and CH2Br2. Atmos. Chem. Phys. 10(5), 2269–2286 (2010)
Liu, Y., Yvon-Lewis, S.A., Hu, L., Salisbury, J.E., O’Hern, J.E.: CHBr3, CH2Br2, and CHClBr2 in U.S. coastal waters during the Gulf of Mexico and East Coast Carbon cruise. J. Geophys. Res.-Oceans 116, C10004 (2011)
Manley, S.L., Goodwin, K., North, W.J.: Laboratory production of bromoform, methylene bromide, and methyl iodide by macaroalgae and distribution in nearshore southern California waters. Limnol. Oceanogr. 37(8), 1652–1659 (1992)
Montzka, S.A., Reimann, S.: Ozone-Depleting Substances (ODSs) and Related Chemicals, in Scientific Assessment of Ozone Depletion: 2010. Global Ozone Research and Monitoring Project Report No. 52, chap. 1. World Meteorological Organization (WMO), Geneva (2011)
Moore, R.M., Webb, M., Tokarczyk, R., Wever, R.: Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures. J. Geophy. Res.-Oceans 101(C9), 20899–20908 (1996)
Nadzir, M.S.M., Phang, S.M., Abas, M.R., Rahman, N., Abdul, A.S.A., Sturges, W.T., Oram, D.E., Mills, G.P., Leedham, E.C., Pyle, J.A., Harris, N.R.P., Robinson, A.D., Ashfold, M.J., Mead, M.I., Latif, M.T., Khan, M.F., Amiruddin, A.M., Banan, N., Hanafiah, M.M.: Bromocarbons in the tropical coastal and open ocean atmosphere during the 2009 Prime Expedition Scientific Cruise (PESC-09). Atmos. Chem. Phys. 14(15), 8137–8148 (2014)
O’Brien, L.M., Harris, N.R.P., Robinson, A.D., Gostlow, B., Warwick, N., Yang, X., Pyle, J.A.: Bromocarbons in the tropical marine boundary layer at the Cape Verde Observatory - measurements and modelling. Atmos. Chem. Phys. 9(22), 9083–9099 (2009)
Ordonez, C., Lamarque, J.F., Tilmes, S., Kinnison, D.E., Atlas, E.L., Blake, D.R., Santos, G.S., Brasseur, G., Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources. Atmos. Chem. Phys. 12(3), 1423–1447 (2012)
Quack, B., Wallace, D.W.R.: Air-sea flux of bromoform: controls, rates, and implications. Glob. Biogeochem. Cycles 17(1), GB001890 (2003)
Sakai, T., Nagai, T., Matsumura, T., Nakazato, M., Sasaoka, M.: Vertical structure of a nonprecipitating cold frontal head as revealed by raman lidar and wind profiler observations. J. Meteorol. Soc. Jpn. 83, 293–304 (2005)
Schauffler, S.M., Atlas, E.L., Blake, D.R., Flocke, F., Lueb, R.A., Lee-Taylor, J.M., Stroud, V., Travnicek, W.: Distributions of brominated organic compounds in the troposphere and lower stratosphere. J. Geophys. Res.-Atmos. 104(D17), 21513–21535 (1999)
Sturges, W.T., Cota, G.F., Buckley, P.T.: Bromoform emission from Arctic ice algae. Nature 358(6388), 660–662 (1992)
Tegtmeier, S., Krueger, K., Quack, B., Atlas, E.L., Pisso, I., Stohl, A., Yang, X.: Emission and transport of bromocarbons: from the West Pacific ocean into the stratosphere. Atmos. Chem. Phys. 12(22), 10633–10648 (2012)
Tokarczyk, R., Moore, R.M.: Production of volatile organohalogens by phytoplankton cultures. Geophys. Res. Lett. 21(4), 285–288 (1994)
Warwick, N.J., Pyle, J.A., Carver, G.D., Yang, X., Savage, N.H., O’Connor, F.M., Cox, R.A.: Global modeling of biogenic bromocarbons. J. Geophys. Res.-Atmos. 111(D24), D007264 (2006)
Yokouchi, Y., Barrie, L.A., Toom, D., Akimoto, H.: The seasonal variation of selected natural and anthropogenic halocarbons in the Arctic troposphere. Atmos. Environ. 30(10–11), 1723–1727 (1996)
Yokouchi, Y., Hasebe, F., Fujiwara, M., Takashima, H., Shiotani, M., Nishi, N., Kanaya, Y., Hashimoto, S., Fraser, P., Toom-Sauntry, D., Mukai, H., Nojiri, Y.: Correlations and emission ratios among bromoform, dibromochloromethane, and dibromomethane in the atmosphere. J. Geophys. Res.-Atmos. 110(D23), D23309 (2005)
Yokouchi, Y., Taguchi, S., Saito, T., Tohjima, Y., Tanimoto, H., Mukai, H.: High frequency measurements of HFCs at a remote site in east Asia and their implications for Chinese emissions. Geophys. Res. Lett. 33(21), L21814 (2006)
Yokouchi, Y., Saito, T., Ooki, A., Mukai, H.: Diurnal and seasonal variations of iodocarbons (CH2ClI, CH2I2, CH3I, and C2H5I) in the marine atmosphere. J. Geophys. Res.-Atmos. 116, D06301 (2011)
Ziska, F., Quack, B., Abrahamsson, K., Archer, S.D., Atlas, E., Bell, T., Butler, J.H., Carpenter, L.J., Jones, C.E., Harris, N.R.P., Hepach, H., Heumann, K.G., Hughes, C., Kuss, J., Krueger, K., Liss, P., Moore, R.M., Orlikowska, A., Raimund, S., Reeves, C.E., Reifenhaeuser, W., Robinson, A.D., Schall, C., Tanhua, T., Tegtmeier, S., Turner, S., Wang, L., Wallace, D., Williams, J., Yamamoto, H., Yvon-Lewis, S., Yokouchi, Y.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide. Atmos. Chem. Phys. 13(17), 8915–8934 (2013)
Acknowledgments
We thank Nobukazu Oda and other staff members of the Global Environmental Forum Foundation (GEFF) for their great help in the maintenance of the analytical systems and stations. This work was partly supported by the Global Environment Fund (Ministry of the Environment of Japan). We also acknowledge the technical assistance of C. Siso and funding in part from the NOAA Climate Program Office’s AC4 program.
Author information
Authors and Affiliations
Corresponding author
Additional information
SI-Natural Halocarbons in the Atmosphere
Rights and permissions
About this article

Cite this article
Yokouchi, Y., Saito, T., Zeng, J. et al. Seasonal variation of bromocarbons at Hateruma Island, Japan: implications for global sources. J Atmos Chem 74, 171–185 (2017). https://doi.org/10.1007/s10874-016-9333-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10874-016-9333-9