
Abstract
Sensitive methods for the determination of arsenate and particulate arsenic (PAs), as well as phosphate and particulate phosphorus (PP), in seawater are described. The method for arsenate and phosphate was established by applying automated liquid waveguide spectrophotometry. Because the reaction time for the formation of the arsenate-molybdate complex is longer than that for the phosphate-molybdate complex, a long Teflon tube submerged in a heating bath was installed in the conventional phosphate flow system. The arsenate was quantified as the difference between absorbances of molybdenum blue dyes with (only phosphate) and without (phosphate + arsenate) arsenate reduction treatment. Contamination was observed in the reagent for arsenate reduction and must be corrected. Linear dynamic ranges up to 1000 nM were confirmed for arsenate and phosphate. The detection limits for arsenate and phosphate were 5 and 4 nM, respectively. Freezing was a reliable sample preservation technique for both arsenate and phosphate. The method for PAs and PP was established by combining conventional persulfate oxidation of PP and the automated liquid waveguide spectrophotometry of arsenate and phosphate. The digestion efficiencies of organic As analogs were >93%. Contamination in the glass fiber filter was negligible. Field tests confirmed that the coefficients of variation (CVs) of 10–19 nM arsenate and 4–151 nM phosphate were 7–20 and 1–25%, respectively, while the CVs of 0.9 nM PAs and 10.2 nM PP were 11 and 4%, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Arsenic (As) exists in both dissolved and particulate forms in seawater (Neff 1997; Henke 2009). Dissolved forms include arsenate, arsenite, and organic compounds, such as methylated As. Among them, arsenate is predominant because it is thermodynamically stable in oxygenated seawater. Arsenate has physicochemical properties similar to those of phosphate. Marine microbial communities uptake arsenate, as well as phosphate, which induce toxic effects due to decoupling energy metabolism. This process involves a transformation from dissolved As to particulate As (PAs). To understand this transformation process, interaction between As and phosphorus (P), and toxic effect on marine ecosystem, information on the size and dynamics of dissolved As and PAs, as well as dissolved P and particulate P (PP), are required.
Several As species in natural water have been determined using a hydride generation method (Andreae 1977; Cutter et al. 1991; Hasegawa et al. 1994), which can be used to detect nanomolar concentrations of As species (Cutter et al. 2001; Cutter and Cutter 2006). Although this method has been widely applied to the study of As biogeochemistry in oceanic water, its application to As and P interaction study is extremely limited because additional measurements of P species are required. A molybdenum blue spectrophotometric method for the determination of arsenate in natural water has also been proposed (Johnson 1971). In this method, arsenate was quantified as the difference between absorbances of the molybdenum blue dyes, with and without arsenate reduction treatment. This method enabled the determination of both arsenate and phosphate, and was useful in biogeochemical studies on As and P, and their interactions. However, there is little application of the molybdenum blue method to oceanic water. Karl and Tien (1992) quantified arsenate concentration in oceanic water using the molybdenum blue method to examine its interference in phosphate measurement. They adopted a magnesium-induced coprecipitation (MAGIC) procedure before spectrophotometric analysis. This procedure concentrated both phosphate and arsenate, and subsequent spectrophotometric analysis detected them at nanomolar concentrations.
Aside from the MAGIC method, sensitive phosphate measurements using 100–250 cm path length liquid waveguide capillary cells (LWCCs) were established (e.g., Zhang and Chi 2002; Hashihama et al. 2009, 2013). The LWCC is composed of quartz tubing with a 550 μm inner diameter, and its outer surface is coated with a low-refractive-index cladding material, Teflon-AF. The sensitive method using the LWCC is automated, has no concentration procedure, and performs almost as well as the MAGIC method (Li and Hansell 2008). Along with phosphate determination, the LWCC method is potentially applicable to the determination of nanomolar arsenate in oceanic water. However, to the best of our knowledge, there has been no report of arsenate determination using the LWCC method.
Furthermore, a sensitive method for the determination of PP in oceanic water was recently established by Ehama et al. (2016). This method was developed by combining the persulfate oxidation of filtered particles (Suzumura 2008) with sensitive spectrophotometry using an LWCC. In theory, the persulfate oxidation can digest not only PP, but also PAs (Hasegawa et al. 1994). Because PAs oxidizes to arsenate in this process, the concentration can be determined by measuring the arsenate concentration using the LWCC method.
In this study, we established LWCC methods for determining arsenate and PAs, as well as phosphate and PP, by modifying the conventional LWCC methods for phosphate (Hashihama et al. 2009, 2013) and PP (Ehama et al. 2016). As the reaction time was longer for the arsenate-molybdate complex than the phosphate-molybdate complex (Johnson 1971), a long Teflon tube in a heating bath was installed in the reaction path of the conventional LWCC method for phosphate. Furthermore, the contamination of As and P in the reagents and filter during sample processing, the effect of sample freezing on arsenate and phosphate determinations, and digestion efficiencies of organic As analogs in the persulfate oxidation were carefully examined. The established methods were applied to the determinations of arsenate and PAs, as well as phosphate and PP, in oceanic waters.
2 Experimental
2.1 Reagents
All reagents in this study were of analytical reagent grade and obtained from Wako Pure Chemical Industries (Osaka, Japan). The pure water used to prepare analytical reagents and stock standard solutions was purified by a reverse osmosis and deionization system (Millipore Auto Pure WEX3 and WR600A, Yamato, Japan), which produced water with a resistance of 18.2 MΩ cm. All plastic and glassware used to store reagents or samples were cleaned carefully using 2% Merck Extran MA 03, and then rinsed with 0.3 M hydrochloric acid and pure water immediately prior to use.
The preparation of analytical reagents for automated analysis was based on that for the automated phosphate analysis, as described by Hansen and Koroleff (1999), with the exception of the ascorbic acid solution (Hashihama et al. 2013), which was prepared by dissolving 0.6 g ascorbic acid in 180 mL pure water and then adding 15 mL acetone and 5 mL of 15% sodium dodecyl sulfate solution. Using acetone and 15% sodium dodecyl sulfate effectively eliminated baseline drift.
Based on the work of Johnson (1971), a reducing reagent (RR) for arsenate reduction was prepared by slowly mixing 20 mL of 1.75 M sulfuric acid into 40 mL of 14% sodium disulfite solution and subsequently mixing with 40 mL of 1.4% sodium thiosulfate solution. The RR was prepared immediately prior to analysis.
The reagents for persulfate oxidation were based on those described by Ehama et al. (2016) after Suzumura (2008). A 0.17 M sodium sulfate (Na2SO4) solution was used to remove dissolved As and P from the filter, and 3% potassium persulfate (K2S2O8) was prepared for use as a digestion reagent.
2.2 Standard, blank, and carrier
Disodium hydrogen arsenate heptahydrate and potassium dihydrogen phosphate were used to prepare 5 and 10 mM stock standard solutions, respectively. For the automated analysis of arsenate and phosphate, working standard solutions were prepared by diluting the stock solution with western North Pacific surface water (WNPS). WNPS was also used as a carrier. Arsenate and phosphate-free seawater (APFS), prepared by the MAGIC procedure (Karl and Tien 1992), was used as a blank.
For the automated analysis of PAs and PP-digested samples, working standards dissolved in 0.5% K2S2O8 and 3% sodium chloride (NaCl) solution (0.5% K2S2O8 + 3% NaCl) were prepared by mixing the arsenate and phosphate standards dissolved in 3.6% NaCl with autoclaved 3% K2S2O8 [5:1 (v:v)]. The 3% NaCl was used as a carrier. The 0.5% K2S2O8 + 3% NaCl was used as a blank (reagent blank, see below).
2.3 Automated analysis of arsenate and phosphate
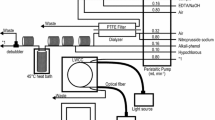
A gas-segmented continuous flow system (AutoAnalyzer II, Technicon, now Seal Analytical, Hampshire, UK) was used for the automated analysis of arsenate and phosphate (Fig. 1). In the detector position, we installed a 100 cm path length LWCC (LWCC-2100, World Precision Instruments, Sarasota, FL, USA), a tungsten fiber optic light source (L7893, Hamamatsu Photonics, Shizuoka, Japan), and a miniature fiber optic spectrometer (USB4000, Ocean Optics, Dunedin, FL, USA). Absorption spectra of arsenate and phosphate were recorded with the spectrometer.

Manifold configuration and flow diagram for gas-segmented continuous-flow analysis of arsenate and phosphate with a 100 cm LWCC. The flow rate of each pumping tube is provided to the right of the peristaltic pump
The present analytical system was based on our analytical system for nanomolar phosphate (Hashihama et al. 2009, 2013), except for a heating bath unit comprising a 2 mm i.d. Teflon tubing (AS ONE, Osaka, Japan) in a temperature-controlled water bath (Isotemp 2320, Thermo Fisher Scientific, Waltham, MA, USA). The heating bath unit was attached for the complete formation of arsenate-molybdate complex (Johnson 1971). To determine optimal conditions for the formation of the arsenate-molybdate complex, we monitored the absorbance of a 100 nM arsenate working standard at reaction temperatures of 25 and 37 °C at reaction times ranging from 35 to 110 min using different lengths of Teflon tubing. These reaction conditions were preliminarily determined based on those of our phosphate system (Hashihama et al. 2009, 2013) and original manual procedure (Johnson 1971).
As arsenate and phosphate were quantified from the absorbances of the molybdenum blue dyes both with and without arsenate reduction, samples with and without RR addition were sequentially injected from a sample suction line of the flow system (Fig. 1). The RR was added to the sample at a volume ratio of 1:10 (RR:sample) (Johnson 1971). The sample mixed with RR was left to stand for 15 min prior to injection. To examine contamination in the RR, absorbance of the RR itself was determined by measuring the RR dissolved in APFS (APFS + RR) at a volume ratio of 1:10.
2.4 Sample processing for PAs and PP
Sample processing for PAs and PP was based on that described by Ehama et al. (2016) after Suzumura (2008). A pre-combusted, acid-washed glass fiber filter (Whatman GF/F, 2.5 cm diameter, Kent, UK) was used to collect particles. Filtration was carried out using an aspirator (A-3S, TOKYORIKAKIKAI, Tokyo, Japan) under vacuum at <0.02 MPa. After filtration, the filter was rinsed with 5 mL of 0.17 M Na2SO4, then dried and placed into a digestion glass bottle (GL32, Duran, Wertheim/Main, Germany). The particles on the filter were digested with 20 mL of 3% K2S2O8 at 120 °C for 30 min using an autoclave (KTS-2322, ALP, Tokyo, Japan). The bottle was shaken before and after autoclaving. The residue in the digested solution was removed using a 0.45 μm syringe filter (Millex-HV, Millipore, Massachusetts, USA). Because >2% K2S2O8 inhibits color development in the sample, the digested solutions were diluted six fold with 3.6% NaCl (i.e., the sample was dissolved in 0.5% K2S2O8 + 3% NaCl). Arsenate and phosphate concentrations in the diluted solution were determined using the LWCC method described above. In the previous LWCC method for PP, pure water was used as a carrier and to prepare the blank and matrices of working standards and samples (Ehama et al. 2016). However, in the present LWCC method for PAs and PP, 3% NaCl was used instead of pure water, because our preliminary experiments showed that using pure water disturbed periodic gas-segmented flow of the analytical system with long Teflon tubing.
Almost complete digestion of oceanic PP during processing has been reported elsewhere (Suzumura 2008), while the digestion efficiency of PAs in this processing was not reported. The digestion efficiency of PAs was confirmed using two methylated As analogs, methylarsonic acid and cacodylic acid (Table 1), which are commonly present in oceanic water (Cutter et al. 2001; Cutter and Cutter 2006). These analogs were dissolved in pure water, and a portion of each solution, containing 12 nmol of As, was dispensed into digestion glass bottles. The analog solutions were gently heated to dryness on a hot plate. The dried samples were digested and analyzed as described above. The final As concentration of each sample was 100 nM, and the digestion efficiency was estimated as a proportion of the absorbance of the digested analogs to the absorbance of the 100 nM arsenate standard. To examine the contamination of arsenate in the analogs, undigested samples were also analyzed.
The absorbances of the procedural blank (GF/F filter + 0.5% K2S2O8 + 3% NaCl) and reagent blank (0.5% K2S2O8 + 3% NaCl) were compared to check As and P contamination in the GF/F filter. The procedural blank was prepared by filtering 1 L of pure water and was processed following the digestion procedure outlined.
2.5 Field sampling
Seawater samples for arsenate and phosphate were collected in oceanic waters of the tropical and subtropical Pacific Ocean (37.5°N–30.0°S) during the KH-11-10 cruise (Dec. 2011–Jan. 2012) and KH-12-3 cruise (Jul.–Aug. 2012) of R/V Hakuho-maru. Sampling was conducted using Teflon-coated 12 L Niskin-X bottles (General Oceanics, Miami, FL, USA) on a CTD-Carousel system (Sea-Bird Electronics, Bellevue, WA, USA). The sample in the Niskin-X bottle was poured into 30 mL polypropylene tube (Sarstedt, Nümbrecht, Germany). For testing the reproducibility of arsenate and phosphate determinations, five replicate samples were collected at depths of 119 m in the central North Pacific, 100 m in the eastern South Pacific, and 5 and 100 m in the western North Pacific (Table 2). All samples were stored frozen at −20 °C during the cruises, and then thawed and analyzed onshore using the automated analytical system for arsenate and phosphate. To examine the effect of sample freezing on arsenate and phosphate determinations, each sample obtained from depths of 5 and 100 m in the western North Pacific was poured into an additional five tubes. These replicate samples were immediately analyzed on board.
Seawater samples for testing the reproducibility of PAs and PP determinations were collected at a depth of 10 m in the western North Pacific during the KH-13-7 cruise of R/V Hakuho-maru (Table 3). The samples were collected using Teflon-coated 12 L Niskin-X bottles on a CTD-Carousel system, and poured into three polycarbonate bottles (Thermo Scientific Nalgene, Rochester, NY, USA). Each sample, with a volume of 1.19 L, was filtered using a GF/F filter. The filters were stored at −20 °C until onshore analysis.
3 Results and discussion
3.1 Arsenate and phosphate determinations
3.1.1 Absorption spectra
Absorption spectra between 500 and 900 nm were obtained by measuring the 100 nM arsenate and 100 nM phosphate working standards (Fig. 2). Baselines were drawn using the WNPS. The arsenate and phosphate spectra derived from the present LWCC method were similar to those from the original method (Johnson 1971). Due to effective detection of the 100 cm LWCC in the range 230–730 nm, absorbances for both arsenate and phosphate highly fluctuated at >730 nm. Because the fluctuation slightly extended to the range 710–730 nm for both arsenate and phosphate, we selected a wavelength of 708 nm for both arsenate and phosphate measurements. This wavelength was also used in our original LWCC method for phosphate (Hashihama et al. 2009, 2013).

Absorption spectra of a arsenate-molybdate complex and b phosphate-molybdate complex measured in the 100 nM arsenate and 100 nM phosphate working standards, respectively
3.1.2 Optimal conditions for the formation of arsenate-molybdate complex
Color development of arsenate-molybdate complex depended on reaction time and temperature (Fig. 3). In Fig. 3, the absorbance of WNPS at each reaction time was set to zero. At 37 °C, the absorbance of 100 nM arsenate increased with time, reaching a plateau at 95 min. The mean absorbance at 95 min was not significantly different from that at 110 min (t test, p > 0.05, n = 3). At 25 °C, the absorbance of 100 nM arsenate gradually increased, but did not reach the maximum absorbance observed at 37 °C. According to these results, we determined that the optimal reaction time and temperature were 95 min and 37 °C, respectively. These conditions were similar to that of the original method (Johnson 1971).

Absorbance of 100 nM arsenate standard at reaction temperatures 25 °C (open circle) and 37 °C (closed circle) at reaction times ranging from 35 to 110 min. Error bars indicates standard deviations (n = 3)
3.1.3 Contamination in reducing reagent
When the absorbance of WNPS was set to zero, the absorbances of APFS + RR and of APFS alone were −0.008 ± 0.003 and −0.014 ± 0.003, respectively [mean ± standard deviation (SD), n = 4]. The absorbance of APFS + RR was significantly higher than that of APFS (t test, p < 0.05), with a difference of 0.006. This difference was ascribed to any contamination in the RR that increased absorbance (e.g., trace phosphate). Because the contamination resulted in significant overestimation of the phosphate concentration, its absorbance had to be corrected during the calculations as shown below.
3.1.4 Output signals
The typical output signals of WNPS with RR (WNPS + RR), APFS, APFS + RR, 100 nM arsenate standard, 100 nM arsenate standard with RR (100 nM arsenate standard + RR), and 100 nM phosphate standard, along with WNPS injections, are shown in Fig. 4. Each suction time was 5 min, which was enough to eliminate the influence of carryover and sample dispersion. Based on sample suction time and the sample line flow rate (1.20 mL min−1, Fig. 1), the sample volumes required were estimated to be 6 mL. In the output signals of Fig. 4, WNPS absorbance was set to zero. The absorbance of WNPS + RR (−0.006 ± 0.001, mean ± SD, n = 3) was slightly lower than that of WNPS. This was ascribed to the reduction of arsenate dissolved in WNPS. The absorbance of the WNPS + RR was not significantly different from that of the 100 nM arsenate standard + RR (t test, p > 0.05, n = 3), indicating that arsenate was completely reduced even at the 100 nM level. As mentioned above, the absorbance of APFS + RR was higher than that of APFS. The absorbance of the 100 nM arsenate standard was significantly lower than that of the 100 nM phosphate standard (t test, p < 0.05, n = 3). These measurements must be made prior to sample measurement to check arsenate reduction efficiency, contamination in the RR, and the absorbance signals of both arsenate and phosphate.

Typical output signals of WNPS + RR, APFS, APFS + RR, 100 nM arsenate standard, 100 nM arsenate standard + RR, and 100 nM phosphate standard along with WNPS injections
3.1.5 Calibration curves and detection limits
Calibration curves were established using working standards of arsenate and phosphate (Fig. 5). In each curve, WPNS absorbance was set to zero. The linear absorbance response to arsenate and phosphate concentrations up to 1000 nM (n = 10) and 40 nM (n = 5) were obtained with strong correlations (r 2 > 0.9971). Detection limits for arsenate and phosphate were estimated as dividing 3SD of the absorbance of APFS + RR (n = 7) by each slope (nM−1) of the calibration curve. The detection limits were 5 and 4 nM for arsenate and phosphate, respectively.

Calibration curves of arsenate up to a 1000 nM and b 40 nM and phosphate up to c 1000 nM and d 40 nM
3.1.6 Calculations of arsenate and phosphate concentrations in natural samples
The absorbance of APFS was set to zero as a blank when calculating arsenate and phosphate concentrations in natural samples. Arsenate and phosphate concentrations (nM) in natural samples were calculated as follows:
where ABSsample is the absorbance of the sample, ABSsample+RR is the absorbance of the sample with RR, ABSAPFS+RR is the absorbance of APFS with RR, 1.1 is the dilution factor with a volume ratio of 11:10 (sample + RR:sample), and SAs and SP are the slopes (nM−1) of the calibration curves of arsenate and phosphate, respectively.
3.1.7 Effect of sample freezing on arsenate and phosphate determinations
Arsenate concentrations in the fresh samples (immediately analyzed onboard) collected at 5 and 100 m in the western North Pacific were 10 ± 2 and 14 ± 1 (mean ± SD, n = 5), respectively, while those in frozen samples collected at 5 and 100 m in the western North Pacific were 11 ± 1 and 15 ± 1 (n = 5), respectively (Table 2). Arsenate concentrations in fresh samples were not significantly different from those in frozen samples (t-test, p > 0.05, n = 5). Phosphate concentrations in the fresh samples at 5 and 100 m in the western North Pacific were 4 ± 1 and 5 ± 1 (n = 5), respectively, while those in the frozen samples at 5 and 100 m in the western North Pacific were 4 ± 1 and 6 ± 1 (n = 5), respectively. As for arsenate concentrations, phosphate concentrations in fresh samples were not significantly different to those in frozen samples (t test, p > 0.05, n = 5). These results indicated that freezing was a reliable method for the preservation of arsenate and phosphate samples.
3.1.8 Concentration and reproducibility of the filed sample
Arsenate and phosphate concentrations in the field samples collected at depths of 10 m in the tropical and subtropical Pacific ranged from 8 to 20 nM and from <4 to 471 nM, respectively (n = 33). The concentration range of arsenate was fairly uniform and much smaller than that of phosphate. The arsenate concentration level was well consistent with that previously observed at the surface (<10 m) of the tropical and subtropical Pacific (12–28 nM, Karl and Tien 1992; Cutter and Cutter 2006). The present arsenate measurement is not influenced by other As species, because the concentrations of arsenite and organic As in oceanic waters are generally at picomolar or subnanomolar level (Cutter et al. 2001; Cutter and Cutter 2006) and far below the detection limit (5 nM) of our analytical system. Thus, the present analytical method could measure reliable arsenate concentrations in oceanic waters.
In contrast, the phosphate concentrations derived from the present analytical method were compared with those derived from the conventional liquid waveguide spectrophotometry of phosphate (Hashihama et al. 2009, 2013) (Fig. 6). Data from the conventional method were obtained from 10 m at the same stations in the KH-11-10 and KH-12-3 cruises and were previously reported in Sato et al. (2013). The phosphate concentrations derived from two methods were not significantly different from each other (all samples: paired t test, p > 0.05, n = 21), even in low concentration range (<50 nM samples: paired t test, p > 0.05, n = 9). The result confirmed that modification of the conventional method did not influence phosphate determination; for example, there was no decomposition of organic P into phosphate due to long reaction time (95 min) and high reaction temperature (37 °C). Furthermore, the conventional phosphate method had no interference from arsenate and silicic acid (Hashihama et al. 2013). This indicates that, in the present phosphate method, arsenate was completely reduced and the interference from silicic acid was negligible.

Phosphate concentrations of the field samples derived from the present and the conventional analytical methods
Mean concentrations of arsenate and phosphate in the replicate samples ranged from 10 to 19 nM and from 4 to 151 nM, respectively (Table 2). Although very low concentrations were observed in several samples, the coefficients of variation (CVs) for replicate samples (n = 5) were less than 20 and 25% for arsenate and phosphate, respectively. These data were good quality, and the present method is suitable for the measurements of oligotrophic samples. However, there remains a concern for arsenate determination in low-arsenate and high-phosphate environments such as nutrient-rich deep water, because high phosphate concentrations at micromolar levels generally have large measurement errors at nanomolar levels, which accompany large errors of nanomolar arsenate through the calculation. In the low-arsenate and high-phosphate environments, the present method is unsuitable, and the arsenate determination should be done using a hydride generation method (Andreae 1977; Cutter et al. 1991; Hasegawa et al. 1994).
3.2 PAs and PP determinations
3.2.1 Digestion efficiencies of organic As analogs
The absorbances of undigested organic As analogs were not significantly different from the blank absorbance (t test, p > 0.05, n = 3), indicating that organic As analogs did not contain inorganic arsenate. A comparison of the absorbances of digested organic As analogs and the arsenate standard revealed that the digestion efficiencies of methylarsonic acid and cacodylic acid were 93 ± 5 and 99 ± 6% (mean ± SD, n = 3), respectively (Table 1). These results implied that the sample processing in this study could digest PAs in the same manner previously observed for PP (Suzumura 2008).
3.2.2 Contamination in glass fiber filter
When the absorbance of 3% NaCl (carrier) was set to zero, the absorbances of the reagent blank and procedural blank were both 0.009 ± 0.001 (mean ± SD, n = 3), with no significant differences (t test, p > 0.05, n = 3). Andreae (1999) described the glass fiber filter potentially containing trace As compounds. However, based on our results, the GF/F filter used here did not contain either As or P compounds. Thus, contamination in the GF/F filter was negligible.
3.2.3 Calculations of PAs and PP concentrations in natural samples
Absorbance of the reagent blank (0.5% K2S2O8 + 3% NaCl) was set to zero when calculating PAs and PP concentrations in natural samples. PAs and PP concentrations (nM) in natural samples were calculated as follows:
where [Arsenatedigested] and [Phoshatedigested] are the concentrations (nM) of arsenate and phosphate in the digested solution, respectively, 0.02 is the volume (L) of 3% K2S2O8, 6 is the dilution factor with a volume ratio of 6:1 (digested solution with 3.6% NaCl:digested solution), and FV is the filtration volume (L). Calculations of [Arsenatedigested] and [Phoshatedigested] were followed by Eqs. (1) and (2), respectively, except for ABSAPFS+RR. In place of the ABSAPFS+RR, the absorbance of RR dissolved in the reagent blank was used here. The slopes (nM−1) of the calibration curves (SAs and SP) used here should be derived from the working standards dissolved in 0.5% K2S2O8 + 3% NaCl, but slope values were not significantly different between the standard matrices of WNPS and 0.5% K2S2O8 + 3% NaCl (t test, p > 0.05, n = 3).
3.2.4 Concentration and reproducibility of the field sample
PAs and PP concentrations in the samples collected at a depth of 10 m in the western North Pacific were 0.9 ± 0.1 and 10.2 ± 0.4 (mean ± SD, n = 3), respectively (Table 3). To the best of our knowledge, these are the first data for both PAs and PP concentrations in oceanic water, although the data were obtained from a single location. The CVs of the PAs and PP samples were 11 and 4%, respectively. The PP concentration with the CV was consistent with those of other oceanic water (Ehama et al. 2016). Indeed, the PAs concentration in oceanic water was very low. If standard spectrophotometry, which generally measures micromolar levels of arsenate, was adopted, 10 to 1000-fold filtration volumes would be required compared with the nanomolar measurement of arsenate. In the present PAs method, which is a technique for determining nanomolar arsenate, accurate measurements with low CV were possible even at practical filtration volumes (1.19 L).
4 Conclusion
The present study established sensitive analytical methods for the determination of arsenate and PAs, as well as phosphate and PP, in seawater using liquid waveguide spectrophotometry. The arsenate measurement was performed by installing a heating bath unit with long Teflon tubing. Careful assessments of contamination in the reagents and filter, sample preservation, and digestion efficiency enabled accurate measurement of natural samples. The present methods are designed for the determination of arsenate, phosphate, PAs, and PP in oceanic water. By applying this new method to extensive observation in oceanic areas, we can gain a better understanding of oceanic As and P dynamics.
References
Andreae MO (1977) Determination of arsenic species in natural waters. Anal Chem 49:820–823
Andreae MO (1999) Arsenic, antimony and germanium, determination of trace elements. In: Grasshoff K, Kremling K, Ehrhardt D (eds) Methods of seawater analysis, 3rd edn. Wiley, Weinheim, pp 274–294
Cutter GA, Cutter LS (2006) Biogeochemistry of arsenic and antimony in the North Pacific Ocean. Geochem Geophys Geosyst 7:Q05M08. doi:10.1029/2005gc001159
Cutter LS, Cutter GA, Diego-McGlone MLCS (1991) Simultaneous determination of inorganic arsenic and antimony species in natural waters using selective hydride generation with gas chromatography/photoionization detection. Anal Chem 63:1138–1142
Cutter GA, Cutter LS, Featherstone AM, Lohrenz SE (2001) Antimony and arsenic biogeochemistry in the western Atlantic Ocean. Deep-Sea Res II 48:2895–2915
Ehama M, Hashihama F, Kinouchi S, Kanda J, Saito H (2016) Sensitive determination of total particulate phosphorus and particulate inorganic phosphorus in seawater using liquid waveguide spectrophotometry. Talanta 153:66–70
Hansen HP, Koroleff F (1999) Determination of nutrients. In: Grasshoff K, Kremling K, Ehrhardt D (eds) Methods of seawater analysis, 3rd edn. Wiley, Weinheim, pp 159–228
Hasegawa H, Sohrin Y, Matsui M, Hojo M, Kawashima M (1994) Speciation of arsenic in natural waters by solvent extraction and hydride generation atomic absorption spectrometry. Anal Chem 66:3252–3547
Hashihama F, Furuya K, Kitajima S, Takeda S, Takemura T, Kanda J (2009) Macro-scale exhaustion of surface phosphate by dinitrogen fixation in the western North Pacific. Geophys Res Lett 36:L03610. doi:10.1029/2008gl036866
Hashihama F, Kinouchi S, Suwa S, Suzumura M, Kanda J (2013) Sensitive determination of enzymatically labile dissolved organic phosphorus and its vertical profiles in the oligotrophic western North Pacific and East China Sea. J Oceanogr 69:357–367
Henke KR (2009) Arsenic in natural environments. In: Henke KR (ed) Arsenic—environmental chemistry, health threats and waste treatment. Wiley, Chichester, pp 69–235
Johnson DL (1971) Simultaneous determination of arsenate and phosphate in natural waters. Environ Sci Technol 5:411–414
Karl DM, Tien G (1992) MAGIC: a sensitive and precise method for measuring dissolved phosphorus in aquatic environments. Limnol Oceanogr 37:105–116
Li QP, Hansell DA (2008) Intercomparison and coupling of magnesium-induced co-precipitation and long-path liquid-waveguide capillary cell techniques for trace analysis of phosphate in seawater. Anal Chim Acta 611:68–72
Neff JM (1997) Ecotoxicology of arsenic in the marine environment. Environ Toxicol Chem 16:917–927
Sato M, Sakuraba R, Hashihama F (2013) Phosphate monoesterase and diesterase activities in the North and South Pacific Ocean. Biogeosciences 10:7677–7688
Suzumura M (2008) Persulfate chemical wet oxidation method for the determination of particulate phosphorus in comparison with a high-temperature dry combustion method. Limnol Oceanogr Methods 6:619–629
Zhang JZ, Chi J (2002) Automated analysis of nanomolar concentrations of phosphate in natural waters with liquid waveguide. Environ Sci Technol 36:1048–1053
Acknowledgements
We are grateful to the officers, crew members, and members of the scientific party of the R/V Hakuho-maru cruises for their cooperation at sea. This work was financially supported by Grant-in-Aid No. 24710004 for Young Scientists (B) from the Japan Society for Promotion of Science and Grant-in Aid No. 24121003 for Scientific Research in Innovative Areas (New Ocean Paradigm on Its Biogeochemistry, Ecosystem and Sustainable Use “NEOPS”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hashihama, F., Suwa, S. & Kanda, J. Liquid waveguide spectrophotometric measurements of arsenate and particulate arsenic, as well as phosphate and particulate phosphorus, in seawater. J Oceanogr 73, 439–447 (2017). https://doi.org/10.1007/s10872-017-0412-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10872-017-0412-6