
Abstract
Two new Zn(II) coordination polymers based on bis(imidazol), namely, [Zn(hba)2(bib)]n(1) and [Zn(NAA)(bib)1.5]n·nNO3−·3nH2O (2) (Hhba = 3-hydroxybenzoic acid, HNAA = 1-naphthyl acetic acid, bib = 1,4-bis(imidazol-1-yl)-butane) have been successfully synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses, elemental analyses, IR, TG and fluorescence spectrums. Complex 1 shows a one-dimensional zigzag chain structure. Complex 2 exhibits two-dimensional network structure. The intermolecular hydrogen bonding and π–π stacking interactions extend the complexes 1 and 2 into supramolecular architectures and play an important role in stabilizing complexes 1 and 2. Furthermore, complex 1 exhibits intense blue luminescence and appears to be a good candidate for novel hybrid inorganic–organic photoactive materials, while no photoluminescent response was observed for complex 2. In addition, we analyzed the Natural Bond Orbitals (NBO) of 1 and 2 using the PBE0/LANL2DZ method in the Gaussian 09 Program. The calculations showed the obvious covalent interaction between the coordinated atoms and Zn(II) ion.
Graphic Abstract
Two new Zn(II) coordination polymers based on bis(imidazol), namely, [Zn(hba)2(bib)]n(1) and [Zn(NAA)(bib)1.5]n·nNO3−·3nH2O (2) have been successfully synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses, elemental analyses, IR, TG and fluorescence spectrum. Complex 1 shows a one-dimensional zigzag chain structure. Complex 2 exhibits two-dimensional network structure. The intermolecular hydrogen bonding and π–π stacking interactions extend the complex 1 and 2 into supramolecular architectures and play an important role in stabilizing complex 1 and 2. In addition, we analyzed Natural Bond Orbital (NBO) of 1 and 2 in using the PBE0/LANL2DZ method built in Gaussian 09 Program. The calculation results showed the obvious covalent interaction between the coordinated atoms and Zn(II) ion.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The design and synthesis of metal–organic coordination polymers relies on the selection of ligands and metal ions and has become a very attractive research field. This is motivated not only by the intriguing structural diversity but also by the demand of applying functional materials into the fields of catalysis, porosity, magnetism, luminescence and nonlinear optics [1,2,3]. In general, grids with various sizes and shapes can be synthesized by choosing suitable single metal ions and organic ligands such as carboxylates and N-donor groups [4,5,6]. Self-assembly is a complex process, highly influenced by many factors, such as the coordination geometry of metal ions, the nature of organic ligands, solvent system, temperature, pH value of the solution, the ratio between metal salt and ligand, the templates and the counter anions [7,8,9,10,11,12,13,14].
These factors have been well-studied, but there are other forces such as hydrogen-bonding, π–π interactions, metal–metal interactions that can greatly influence the supramolecular topology and its dimensionality [15,16,17]. These considerations made us investigate new coordination structures with 3-hydroxybenzoic acid (Hhba), 1-naphthyl acetic acid (HNAA) and bridging ligands (bib). In this manuscript, we report the syntheses, crystal structures, IR, fluorescence, and TG properties of two new complexes, [Zn(hba)2(bib)]n(1) and [Zn(NAA)(bib)1.5]n·nNO3−·3nH2O (2). Complex 1 exhibits intense blue luminescence and appears to be good candidate for novel hybrid inorganic–organic photoactive materials, while no photoluminescent response was observed for complex 2. We also analyzed the Natural Bond Orbitals (NBO) using the PBE0/LANL2DZ method in the Gaussian 09 Program.
Experimental Section
General Procedures
All solvents and chemicals were commercial reagents and used without further purification. Elemental analyses (carbon, hydrogen, and nitrogen) were performed with a Vario EL(III) Elemental Analyzer. IR spectra (4000–400 cm−1) were measured from KBr pellet on a Nicolet 6700 FT-IR spectrometer. TG studies were carried on a STA7300 analyzer under nitrogen at a heating rate of 10 °C min−1. The fluorescent studies were carried out on a computer-controlled JY Fluoro-Max-3 spectrometer at room temperature. The crystal structure was determined using a Bruker D8 QUEST CMOS diffractometer. Power X-ray diffraction (PXRD) patterns were obtained on Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å) at room temperature.
Syntheses
[Zn(hba)2(bib)]n(1)
A mixture of Zn(OAc)2·2H2O (0.044 g, 0.2 mmol), Hhba (0.014 g, 0.1 mmol), bib (0.019 g, 0.1 mmol) and 10 mL H2O was stirred for 15 min at room temperature, then sealed in a 20 mL Teflon-lined stainless steel vessel, and heated to 110 °C for 5 days, followed by slow cooling (a descent rate of 5 °C h−1) to room temperature. Colorless prism-shaped crystals were obtained. Yield of 26% (based on bib). Anal. Calcd. For C24H22N4O6Zn: C, 54.61; H, 4.20; N, 10.61. Found C, 54.03; H, 3.94; N, 10.08. IR (cm−1): 3274(w), 1630(w), 1576(s), 1450(m), 1387(m), 1230(m), 1216(w), 1081(m), 936(w), 892(w), 785(w), 713(w), 641(w), 462(w).
[Zn(NAA)(bib)1.5]n·nNO3−·3nH2O (2)
A mixture of Zn(NO3)2·6H2O (0.029 g, 0.1 mmol), HNAA (0.019 g, 0.1 mmol), bib (0.019 g, 0.1 mmol), C2H5OH (5 mL) and 5 mL H2O was stirred for 15 min at room temperature, then sealed in a 20 mL Teflon-lined stainless steel vessel, and heated to 120 °C for 5 days, followed by slow cooling (a descent rate of 5 °C h−1) to room temperature. Colorless prism-shaped crystals were obtained. Yield of 27% (based on Zn). Anal. Calcd. For C27H36N7O8Zn: C, 49.74; H, 5.57; N, 15.04. Found C, 49.01; H, 5.23; N, 14.67. IR (cm−1): 3423(w), 3127(w), 1586(m), 1528(m), 1390(s), 1231(m), 1094(s), 949(w), 797(w), 659(w).
Structure Determinations
The X-ray diffraction data collection was performed on two single crystals with the dimensions of 0.14 × 0.11 × 0.08 mm3 for 1 and 0.15 × 0.13 × 0.09 mm3 for 2 respectively, were collected at 293(2) K on a Bruker D8 QUEST CMOS diffractometer with Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct method and refined by full-matrix least-squares on F2 using the SHELXL-97 program [18, 19]. The non-hydrogen atoms of the complexes were refined with anisotropic temperature parameters. The hydrogen atoms were positioned geometrically and refined using a riding model. Crystallographic parameters and the data collection statistics for structures 1 and 2 are shown in Table 1. Selected bond lengths and angles are given in Table 2. Hydrogen bond parameters of 1 and 2 are listed in Table 3. Further crystallographic parameters have been deposited with the Cambridge Crystallographic Data Centre (no. 1535330 (1), 1567036 (2); deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/data_request/cif).
Results and Discussion
Structure Description
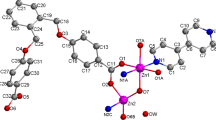
Complex 1 exhibits a one-dimensional zigzag chain structure. The asymmetric unit of 1 contains one crystallographically unique Zn(II) ion, two hba ligands and one bib molecule. As shown in Fig. 1, the Zn(1) ion is four-coordinated and displays a distorted tetrahedral {ZnN2O2} geometry completed by two carboxylate oxygen atoms from two different hba ligands and two nitrogen donors from two flexible bib molecules. The Zn–O bond distances are in the range of 1.9384(18)–1.9432(18) Å, and those of Zn–N are between 1.999(2) and 2.012(2) Å. The N(O)–Zn–O(N) angles fall in the 101.80(9)°–119.27(8)° range.

View of the asymmetric unit of the complex 1. Hydrogen atoms are omitted for clarity
It is interesting to note that the deprotonated carboxylates of the hba anion coordinate Zn(II) in a monodentate bridging mode and the bib ligands feature a trans-conformation bridging mode with a dihedral angle between the two imidazole rings of 0°. The bib ligands link adjacent Zn(II) ions to generate a 1D zigzag chain structure with a Zn···Zn distance of 12.218 and 13.478 Å (Fig. 2). These neighboring chains are linked via C–H···O hydrogen bonding interactions (between C atoms of hba, bib ligands and carboxyl O, nitro O atoms), forming a 2D network structure (Fig. 3). In addition, π–π interactions are observed between the imidazole rings of bib ligands. The centroid-to-centroid distance between adjacent rings is 3.6246(17) Å for N1, C15, N2, C17, C16 and N3, C20, N4, C22, C21 (symmetry codes: 1/2 − x, − 1/2 + y, 1/2 − z) imidazole rings. The plane-to-plane distance is 3.5355(12) Å for N1, C15, N2, C17, C16 and N3, C20, N4, C22, C21 (symmetry codes: 1/2 − x, − 1/2 + y, 1/2 − z) imidazole rings. Thus, through C–H···O hydrogen bonding and π–π interactions, a 3D supramolecular architecture is achieved.

View of the one-dimensional zigzag chain of complex 1

View of the 2D network structure of complex 1 formed by C–H···O hydrogen bonding
Complex 2 exhibits a two-dimensional network architecture. Its asymmetric unit consists of one Zn(II) ion, one NAA ligand, one and a half bib molecules, one counterion nitrate and three crystal water molecules. As depicted in Fig. 4, the four-coordinated Zn(1) ion displays a distorted tetrahedral {ZnON3} geometry coordinating one carboxylate oxygen atom from the NAA ligand and three nitrogen donors from three flexible bib molecules. The Zn–O distance is 1.9518(19) Å, the Zn–N distances are between 1.993(2) and 2.037(2) Å. In 2, the NAA ligand features a monodentate bridging mode, while the bib ligands are in trans-conformation bridging mode with dihedral angles between the two imidazole rings of 0°. The bib ligands link neighboring Zn(II) ions to produce a 2D network architecture with seventy-two membered metalla-macrocycle (Fig. 5). The extended structure of complex 2 includes two kinds of O–H···O and C–H···O hydrogen bonding interactions between carboxylate oxygen atoms, coordinated water molecules and carbon atoms (Table 3). Moreover, π–π interactions between benzene rings of the NAA ligands are observed. The centroid-to-centroid distances between adjacent rings is 3.690(2) Å for C3, C4, C5, C6, C7, C12 and C3′, C4′, C5′, C6′, C7′, C12′ (2 − x, − y, 1 − z) benzene rings with the plane-to-plane distance of 3.4751(17) Å and 3.841(2) Å for C3, C4, C5, C6, C7, C12 and C7, C8, C9, C10, C11, C12 (2 − x, − y, 1 − z) benzene rings with their plane-to-plane distance to be 3.4767(16) Å. Thus, through hydrogen bonds and π–π interactions, the two-dimensional network is further expanded into a three-dimensional supramolecular architecture.

View of the asymmetric unit of the complex 2. Hydrogen atoms are omitted for clarity

View of the 2D network structure of complex 2
IR Analysis of Complexes 1 and 2
In 1, the C–N absorption peaks of imidazole can be observed at 1230 cm−1. Asymmetric and symmetric COO− stretching modes of the lattice hba anion were evidenced by very strong, slightly broadened bands at 1576 and 1387 cm−1 [20], which is consistent with the results of X-ray analysis.
The IR spectrum of 2 shows a broad absorption band at 3423 cm−1 corresponding to the H···O stretching of crystal water molecules in the complex. The C–N absorption peaks of imidazole can be observed at 1231 cm−1. Asymmetric and symmetric COO− stretching modes of the lattice NAA anion were evidenced by very strong, slightly broadened bands at 1586 and 1390 cm−1 [20], which is consistent with the results of X-ray analysis.
Thermal Stability and Powder X-Ray Diffraction (PXRD)
To determine the thermal stability of complexes 1 and 2, their thermal behaviors were investigated under a nitrogen atmosphere by thermogravimetric analysis (TG). As depicted in Fig. 6, the TG curve of 1 shows that the complex is stable up to 230 °C, and then decomposes upon further heating. The TG curve of 2 (Fig. 7) reveals that three crystal water molecules are released between 65 and 320 °C (Obsd. 7.8%; Calcd. 8.3%), and the dehydrated solid begins to decompose at 361 °C. The powder XRD patterns for the as-synthesized bulk material closely match the simulated ones from the single-crystal structure analysis, which is evidence that we obtain a pure solid-state phase (Fig. 8).

TG curve of complex 1

TG curve of complex 2

PXRD patterns of complexes 1 and 2 at room temperature; red simulation and black experimental data (Color figure online)
Photoluminescent Properties
Metal–organic coordination polymers, especially d10 metal centers, such as AgI, AuI, ZnII and CdII, and conjugated organic linkers have been researched because of their fluorescent properties and potential applications as fluorescent-emitting materials, chemical sensors and electroluminescent displays [21]. The solid-state photoluminescent emission spectrum of complex 1 was investigated in at room temperature (Fig. 9). When excited at 330 nm, and complex 1 exhibits intense blue luminescence and shows an emission peak at 367 nm. The free ligands Hhba and bib show photoluminescence with the emission maxima at 410 nm and 438 nm, respectively (λex = 315 nm), which can be assigned to intraligand (π* → π) transitions [22]. Thus, the emission band of complex 1 could be assigned to the emission of ligand-to-ligand charge transfer [23, 24]. Owing to the strong fluorescent intensity, 1 appears to be a good candidate for novel hybrid inorganic–organic photoactive materials.

Solid-state emission spectrum of 1 at room temperature
However, no photoluminescent response was observed for complex 2.
Theoretical Calculations
The presented calculations were performed with the Gaussian 09 program [25]. Experimental data of the complexes 1 and 2 provided the start geometries of the molecular structures for calculation. We analyzed the NBO by DFT [26] with the PBE0 [27,28,29,30] hybrid functional and the LANL2DZ basis set [31].
The selected natural atomic charges, natural electron configuration, Wiberg bond indices and NBO bond orders (a.u) for the compound 1 are listed in Table 4 with the electronic configurations of Zn(II) ion, nitrogen, and oxygen atoms being 4s0.313d9.974p0.39, 2s1.372p4.21–4.24 and 2s1.66–1.682p5.11–5.17, respectively. We infer that the Zn(II) ion coordination with nitrogen and oxygen atoms is mainly on 3d, 4s, and 4p orbitals. Both nitrogen and oxygen atoms form coordination bonds with Zn(II) ion using 2s and 2p orbitals and the Zn(II) ion gained electrons from two nitrogen atoms of bib ligand and two oxygen atoms of hba ligands [32, 33]. On the basis of valence-bond theory, the atomic net charge distribution and the NBO bond orders of the compound 1 (Table 4) exhibits the obvious covalent interaction between Zn(II) and it coordination partners.
As can be seen from Fig. 10, lowest unoccupied molecular orbital (LUMO) is mainly composed of bib molecule, highest occupied molecular orbital (HOMO) mainly consists of bib ligand too. So, the ILCT (intraligand charge-transfer) may be inferred from some contours of molecular orbital of complex 1.

Frontier molecular orbitals of the complex 1
The selected natural atomic charges and natural electron configuration for the complex 2 is shown in Table 4. It is indicated that the electronic configurations of Zn(II) ion, N and O atoms are 4s0.313d9.974p0.39, 2s1.682p5.14 and 2s1.362p4.24–4.28, respectively. Based on the above results, one can conclude that the Zn(II) ion coordination with N and O atoms is mainly on 3d, 4s, and 4p orbitals. N atoms form coordination bonds with Zn(II) ion using 2s and 2p orbitals. All O atoms supply electrons of 2s and 2p to Zn(II) ion and form the coordination bonds. Therefore, the Zn(II) ion obtained some electrons from three N atoms of bib ligand, one O atom of NAA ligand [32, 33]. Thus, according to valence-bond theory, the atomic net charge distribution and the NBO bond orders of the complex 2 (Table 4) shows the obvious covalent interaction between the coordinated atoms and Zn(II) ion. The differences of the NBO bond orders for Zn–O and Zn–N bonds make their bond lengths be different [33], which is in good agreement with the X-ray crystal structural data of complex 2.
As can be seen from Fig. 11, both the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) are mainly located on the bib ligand, and LLCT (ligand-to-ligand charge-transfer) may be inferred from some contours of the molecular orbital of complex 2.

Frontier molecular orbitals of the complex 2
Conclusions
We have presented two new Zn complexes with hba/NAA and bib ligands. The hba and NAA ligands adopt monodentate bridging modes, while the bib ligands exhibit trans-conformation bridging mode and link neighboring Zn(II) ions to generate a 1D zigzag chain in 1 and a 2D network in 2. Furthermore, 3D supramolecular architectures are formed via hydrogen bonding and π–π interactions. A NBO study confirms the obvious covalent interaction between the Zn(II) ions and the coordinated O and N atoms.
References
Ma L, Wu CD, Wanderley MM, Lin W (2010) Angew Chem Int Ed 49:8244
Zhang ZQ, Huang RD, Xu YQ, Hu CW (2008) Chem J Chin Univ 29:1528
Liu GZ, Xin LY, Wang LY (2011) CrystEngComm 13:3013
Li GF, Wang YN, Wang QW, Li XM, Ji JY, Pan YR (2015) Chin J Inorg Chem 31:183
Sorace L, Benellib C, Gatteschi D (2011) Chem Soc Rev 40:3092
Jiang DY, Sui W, Li XM, Liu B, Wang QW, Pan YR (2016) Chin J Struct Chem 35:505
Dechambenoit P, Long JR (2011) Chem Soc Rev 40:3249
Hong MC, Zhao YJ, Su WP, Cao R, Fujita M, Zhou ZY, Chan ASC (2000) J Am Chem Soc 122:4819
Abrahams BF, Batten SR, Grannas MJ, Hamit H, Hoskins BF, Robson R (1999) Angew Chem Int Ed 38:1475
Bu XH, Chen W, Lu SL, Zhang RH, Liao DZ, Bu WM, Shionoya M, Brisse F, Ribas J (2001) Angew Chem Int Ed 40:3201
Qi Y, Che YX, Zheng JM (2008) CrystEngComm 10:1137
Aakeröy CB, Champness NR, Janiak C (2010) CrystEngComm 12:22
Kasai K, Aoyagi M, Fujita M (2000) J Am Chem Soc 122:2140
Du M, Bu XH, Guo YM, Ribas J, Diaz C (2002) Chem Commun 21:2550
Li XM, Pan YR, Ji JY, Niu YL, Niu QW (2014) J Inorg Organomet Polym 24:836
Pan YR, Sun M, Li XM (2015) Chin J Struct Chem 34:576
Tang YZ, Zhou M, Huang J, Tan YH, Wu JS, Wen HR (2013) Inorg Chem 52:1679
Sheldrick GM (1997) SHELXS-97, programs for X-ray crystal structure solution. University of Göttingen, Göttingen
Sheldrick GM (1997) SHELXL-97, programs for X-ray crystal structure refinement. University of Göttingen, Göttingen
Devereux M, Shea DO, Kellett A, McCann M, Walsh M, Egan D, Deegan C, Kedziora E, Rosair G, Müller-Bunz H (2007) Inorg Biochem 101:881
Kreno LE, Leong K, Farha OK, Allendorf M, Duyne RPV, Hupp JT (2012) Chem Rev 112:1105
Lin JD, Long XF, Lin P, Du SW (2010) Cryst Growth Des 10:146
Mohamed GG, El-Gamel NEA (2004) Spectrochim Acta A 60:3141
Li GL, Liu GZ, Huang LL, Li L, Zhang X (2014) J Inorg Organomet Polym 24:617
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian Inc., Wallingford
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, Oxford
Ernzerhof M, Scuseria GE (1999) J Chem Phys 110:5029
Adamo C, Barone VJ (1999) Chem Phys 110:6158
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Perdew JP, Burke K, Ernzerhof M (1997) Phys Rev Lett 78:1396
Dunning TH, Hay PJ Jr (1976) In: Schaefer HF III (ed) Modern theoretical chemistry. Plenum, New York, p 1
Wang L, Zhao J, Ni L, Yao J (2012) J Inorg Gen Chem 638:224
Li ZP, Xing YH, Zhang YH (2009) Acta Phys Chim Sin 25:741
Acknowledgements
This work was supported by the Science and Technology Development Project of Jilin Provincial Science & Technology Department (Grant No. 201205080) and the Science and Technology Research Projects of the Education Department of Jilin Province (Grant No. 2013.384). Program supports from State Key Laboratory of Theoretical and Computational Chemistry of Tonghua Normal University are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, XM., Wang, ZT., Valtchev, V. et al. Syntheses, Crystal Structures and NBO Calculation of Two New Zinc(II) Coordination Polymers. J Chem Crystallogr 50, 155–163 (2020). https://doi.org/10.1007/s10870-019-00807-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-019-00807-3