
Abstract
The crystal structures of two compounds,ethyl-(E)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate and ethyl-(Z)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate were determined from single-crystal X-ray diffraction and characterized by FT-IR, 1HNMR and 13CNMR spectroscopy. The quantum theoretical calculations for two structures were performed by density functional theory (DFT/B3LYP/6-311+G*). From the optimized structure, geometric parameters were obtained and experimental measurements were compared with the calculated data. Frontier molecular orbitals, total density of states, molecular electrostatic potential, molecular properties, thermodynamic parameters and NBO analysis for E and Z isomers were investigated by theoretical calculations.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acrylic compounds have great reactivity as electrophiles towards nucleophiles, particularly in the preparation of natural and biologically active compounds [1, 2], therefore they are good Michael acceptors [3]. Acrylates are salts, esters, and conjugate bases of acrylic acid and its derivatives. They are also known as propenoates. Acrylates easily form polymers because the double bonds are very reactive. These polymers play an important role in military and commercial products [4]. Compounds containing alkyl acrylate skeletons have significant importance in material science [5]. In fact, acrylates are mainly used to make plastic items that start as powders and liquids that are mixed together to make a paste that can be formed into any shape and then hardened. Because of their importance in industry, substituted acrylates have recently received a great deal of attention [6, 7]. In this paper, we report the synthesis, characterization and crystal structures of ethyl-(E)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate and ethyl-(Z)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate (see Fig. 5). In recent years, computational chemistry has become an important tool for chemists and a well-accepted complement for experimental chemistry [8,9,10,11,12]. Density functional theory (DFT) method has become a major tool in the methodological arsenal of computational organic chemists. In the present work, we also investigate the energetic and structural properties of the two structures determined in solid state, using DFT calculations. The optimized geometry, frontier molecular orbitals (FMO), detail of quantum molecular descriptors, molecular electrostatic potential (MEP), chemical tensors, natural charge and NBO analysis were calculated.
Experimental
Physical Techniques and Materials
All reagents and solvents for synthesis and spectroscopic studies were commercially available and used as received without further purification. 1H NMR and 13C NMR spectra were measured for samples in CDCl3 with a BRUKER DRX-250 AVANCE spectrometer at 250 and 62.5 MHz respectively, using TMS as internal standard. The infrared (IR) spectra were recorded on a Mattson-1000 FT spectrometer using KBr pellets. Melting points were measured on an Electrothermal 9100 apparatus.
General Procedure for the Synthesis of E and Z Isomers
A solution of triphenylphosphine (1.0 mmol, 0.262 g) and isatin (1.0 mmol) in dry dichloromethane (10 ml) was placed in a three-necked flask in an ice-water bath. Then, a solution of acetylenic esters (1.0 mmol) in dry dichloromethane (4 ml) was added dropwise and stirred for 5 min. After 5 min, the mixture was cooled to room temperature and then stirred for 15 min until the reaction was completed as monitored by TLC (n-hexane/EtOAc; 2:1) analysis. The solvent was removed under reduced pressure and the residue was purified by column chromatography with silica gel 60 HF-254 using petroleum ether/EtOAc (10:2) as eluent.
The single crystals of E and Z isomers were obtained from slow evaporation of their dichloromethane/light petroleum ether (1:2) solution (20–25 °C). The single crystals were filtered off, washed with a cold mixture of dichloromethane/light petroleum ether (1:2) and dried at room temperature.
The products were characterized by spectral data (IR, 1H NMR and 13C NMR). The physical and spectral data for the products are given below:
Ethyl-(E)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate
Orange Crystals
Yield: 46%. Mp: 106–108 °C. IR (KBr): ν = 3061(CH, arom), 2984 (CH, alipha), 1746, 1723, 1615 (3C=O), 1469 (C=C, alkene) cm−1, 1H NMR (CDCl3) δ H (ppm): 1.1 (t, 3 H, 3JHH=7.0 Hz, CH3), 4.2 (q, 2 H, 3JHH=7.0 Hz, OCH2), 7.1–7.7 (m, 9 H, arom), 5.3 (s, 1 H, =CH). 13C NMR (CDCl3) δ C (ppm): 14 (CH3), 62.2 (OCH2), 132.5 (=CH), 111.2, 118.2, 120, 122.5, 123.2, 124.4, 125, 128.3, 129.6, 129.8, 138.6, 141.2, 151.2 (12 C of aromatic and =C–N), 158 (C=O of amide), 163 (C=O of ester), 182.4 (C=O of ketone).
Ethyl-(Z)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate
Red Crystals
Yield:54%. m.p. 168–170 °C. IR (KBr): ν = 3091 (CH, arom), 2923 (CH, alipha), 1746, 1746, 1615 (3C=O), 1469 (C=C, alkene) cm− 1, 1H NMR (CDCl3) δ H (ppm): 1.3 (t, 3 H, 3JHH=7.0,CH3), 4.3 (q, 2 H, 3JHH=7.0 Hz,OCH2), 6.6–8 (m, 9 H, arom), 8.1 (s, 1 H, =CH). 13CNMR (CDCl3) δ C (ppm): 14.2 (CH3), 62.3 (OCH2), 131.6 (=CH), 111.5, 118.2, 121.1, 124.4, 125.8, 128.2, 129.2, 129.8, 131.2, 135.8, 138.7, 142.6, 150.6 (12 C of aromatic and =C–N), 158 (C=O of amide), 163.00 (C=O of ester), 181.9 (C=O) of ketone).
X-Ray Crystallography
The crystallographic measurements of E and Z isomers were performed on a κ-geometry KUMA KM4CCD automated four-circle diffractometer with graphite-monochromatized MoKα radiation (λ = 0.71073 Å). The data for the crystals were collected at 100(2) K by using the Oxford-Cryosystems cooler. Data were corrected for Lorentz and polarization effects. Data collection, cell refinement, data reduction, and analysis were carried out with CrysAlis CCD and CrysAlis RED, respectively [13]. The structures were solved by direct methods with the SHELXS-97 program [14], and refined by a full-matrix least-squares technique with SHELXL-2013 (for E isomer)and SHELXL-2014(for Z isomer) [15]. Anisotropic thermal parameters were employed for non-H atoms. All H atoms were found in difference Fourier maps. In the final refinement cycles, they were repositioned in their calculated positions and refined using a riding model, with C–H=0.95–0.99 Å, and with U iso(H) =1.2U eq(C) for CH and CH2 or 1.5U eq(C) for CH3. Figures were made with the Diamond program [16]. Details for the data collection and the structures refinements are given in Table 1 and the crystallographic information files (CIFs) deposited with The Cambridge Crystallographic Data Centre (http://www.ccdc.cam.ac.uk/; deposition numbers CCDC-1410473 for E and 1410474 for Z isomer)and provided as Electronic Supplementary Material.
Computational Details
In this work, we have carried out quantum theoretical calculations and have optimized structures of the E and Z isomers of ethyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate, using B3LYP/6-311+G* level (DFT) [17,18,19] by the Gaussian 03 W program package [20]. The electronic properties such as EHOMO, ELUMO, HOMO–LUMO energy gap (∆E), EHOMO−1, ELUMO+1, natural charges, molecular properties, dipole moment (μ D ) and point group were detected. The optimized molecular structure, HOMO and LUMO surfaces were visualized using the GaussView 03 program [21].We also studied the thermodynamic parameters of molecules using the B3LYP/6-311+G* level, and obtained E0 (sum of the electronic and the zero-point energies), enthalpy H (sum of the electronic and the thermal enthalpy), Gibbs free energy G (sum of the electronic and the thermal free energy), entropy S and Cv (constant volume molar heat capacity The electronic structure of E and Z isomers were studied by using Natural Bond Orbital (NBO) analyses at the same level in order to understand various second-order interactions between the filled orbitals of one subsystem and vacant orbitals of another subsystem, which is a measure of the inter-molecular delocalization or hyper conjugation [22, 23].
Result and Discussion
The experimental FT-IR spectra of E and Z isomers are given in Fig. 1 and are compared with calculated FT-IR spectra. Their X-ray structures are also shown.

a Experimental (top) and calculated (bottom) FT-IR spectra of E and Z isomers (using the B3LYP/6-311+G* level). b X-ray structures with atom-numbering scheme of E and Z isomers (displacement ellipsoids shown at the 50% probability level)
Crystal Structures of E and Z Isomers of Ethyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate
The molecules of ethyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate present in the two crystals reported here, adopt respectively E and Z geometry around the C=C double bond, which is reflected in the N(1)–C(4)–C(5)–C(6) torsion angles (Table 2). As shown in Fig. 1, the overall geometry of the E and Z isomers, resemble those observed in related compounds, i.e. derivatives with 2-N-substituted ethyl 3-phenylprop-2-enoate moiety, reported by us previously [24,25,26,27], and deposited with the Cambridge Structural Database, CSD ver. 5.38 update Feb. 2017. A total of 18 compounds were found, from which only 10 examplesFootnote 1contain pure 3-C6H5ring and only two are E isomers [28]. However, closer inspection reveals some significant differences. The carbonyl atom O(2) of the ester group is antiperiplanar to the atom C(5) in the Z isomer (see the O(2)–C(3)–C(4)–C(5) torsion angle in Table 2), while it is anticlinal in the E isomer (135.79(11)°). It is noteworthy that in all the structurally related compounds reported to date, C=O and C=C bonds were approximately coplanar (ap rather than sp), regardless of the N-substituent in 2-position and the (2E)/(2Z) isomerism. In the title compound, the phenyl ring is twisted relative to the C=C bond to a similar extent in E and Z isomer (at about 55° and 35°, respectively), which is shown by the C(4)–C(5)–C(6)–C(7) torsion angles. Orientation of the 1 H-indole-2,3-dione moiety in relation to the rest of the molecule may be defined by the C(19)–N(1)–C(4)–C(5) torsion angle. For comparison of the geometry of both isomers see Fig. 2.

Comparison of the geometry of E (yellow) and Z (grey) isomers. The common reference points are labeled atoms
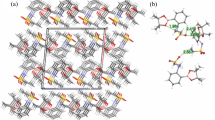
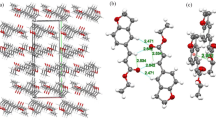
As shown in Figs. 3 and 4, the packing diagrams of the crystals of both E and Z isomers are dominated by C–H⋯O contacts. In the crystal of the E isomer, close carbonyl⋯πcontacts are also found (Fig. 3; O⋯centroid distance of 2.945(2) Å, perpendicular O⋯centroid distance of 2.749 Å, and C=O⋯centroid angle of 142.09(7)°).These link the molecules of E isomer into ribbons along the b-axis, which further interact via C(10)–H(10)⋯π contacts with a H⋯π distance of 2.92 Å and a C–H⋯π angle of 134°. A set of C–H⋯O contacts and π⋯π stacking interactions present in the crystal of Z isomer gives rise to a three-dimensional network, as shown in Fig. 4.

Intermolecular contacts (see Table S1) within a ribbon along the b-axis in the crystal of E isomer. Hydrogen bonds, closest π⋯π and Ocarbonyl⋯π contactsare shown with dashed and blue/red dotted lines, respectively. H atoms not involved in C–H⋯O interactions are omitted for clarity. Symmetry codes: (i) − x + 1, y − 1/2, −z + 1/2; (ii) x, y − 1, z

Arrangement of the molecules of the Z isomer in the crystal lattice: C–H···O interactions (dashed lines) and π⋯π stacking (dotted lines) linking the molecules along the (a) b- and c-axis, b along the a-axis; see Table S2. H atoms not involved in C–H⋯O interactions are omitted for clarity. Symmetry codes: (i) x − 1/2, −y + 3/2, z − 1/2; (iii) x − 1/2, −y + 3/2, z + 1/2; (v) − x + 2, −y + 1, −z + 2; (vi) x − 1, y, z; (#) − x + 3/2, y − 1/2, −z + 3/2; (@)x, y, z − 1
Optimized Geometry
The optimization of the structures of the title molecules were performed at the B3LYP/6-311+G* level (see Fig. 5).The selected bond lengths and bond angles of the crystal structures and the theoretical parameters are listed in Table 2.As can be seen, the calculated parameters show good agreement with the experimental data and can be used as foundation to calculate the other parameters for the title compounds. The slight differences between the calculated and experimental data may result from the intermolecular interactions present in the crystal lattice, which are not taken into account by the computational methods.

The theoretical geometric structure of E and Z (optimized with B3LYP/6-311+G* level)
Electronic Properties
Quantum chemical methods are important to obtain information about molecular structure and electrochemical behavior. Frontier molecular orbitals (FMO) analyses were performed for the E and Z isomers using the B3LYP/6-311+G* level [29].FMO results such asenergy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO) [30], EHOMO−1, ELUMO+1 and HOMO–LUMO energy gap (Eg) for the E and Z isomersare summarized in Table 3. Also, the HOMO can act as an electron donor and the LUMO can act as the electron acceptor. A higher EHOMO for the molecule indicates a higher electron-donating ability to an appropriate acceptor molecule with a low-energy empty molecular orbital [31].As shown in Fig. 6 and Table 3, EHOMO values of compounds Z and E are − 6.69 and − 6.52 eV, respectively. As can be seen from values of EHOMO, isomer E has the higher EHOMO rather than isomer Z, therefore isomer E has a higher electron-donating ability to an appropriate acceptor molecule. As seen in Fig. 6, charge transfer is taking place within E and Z. The graphic representation of HOMO and LUMO orbitals show that the HOMO orbital of isomer E is localized mainly on the isatin ring, the phenyl ring and the C=C moiety, whereas the LUMO orbital is focused mainly on the isatin ring. The HOMO→LUMO transition implies an electron density transfer to the isatin ring. According to graphic representation of the HOMO and LUMO orbitals of isomer Z, the HOMO orbital of isomer Z is localized mainly on the isatin ring and the C=C moiety, while the LUMO orbital is focused mainly on the isatin ring. The HOMO→LUMO transition implies an electron density transfer to the isatin ring. The electronic structures of isomers E and Z were also studied using total densities of states (DOSs) [32].The DOS plot shows a population analysis per orbital and demonstrates a simple view of the character of the molecular orbitals in a certain energy range [33]. According to Fig. 6, the DOS analysis indicates calculated energy gaps (Eg) for molecule E and Z. A large energy gap implies high stability for the molecule. According to Fig. 6, the HOMO–LUMO energy gap (Eg) value for Z isomer (3.79 eV) is higher than that for structure E (3.55 eV). Therefore Z is less reactive rather than E.

Calculated Frontier molecular orbitals for E and Z isomers (Eg: energy gap between LUMO and HOMO) and Calculated DOS plots for E and Z (using the B3LYP/6-311+G*)
Several quantum molecular descriptors for E and Z isomers are summarized in Table 3. I is the ionization potential (I= −E HOMO ) and A is the electron affinity (A= −E LUMO ) [34]. The chemical hardness (η= (I−A)/2) is an important property to measure the molecular stability and reactivity [35]. A hard molecule has a large energy gap (Eg) and a soft molecule has a small energy gap (Eg) [36]. The chemical hardness (η) value for E and Z is 1.77 and 1.89 eV, respectively. Therefore Z is a hard molecule and less reactive with a high energy gap (Eg = 3.79 eV and η = 1.89 eV), whereas E is a soft molecule and more reactive with a low energy gap value (Eg = 3.55 eV and η = 1.77 eV). The electronic chemical potential [µ=−(I + A)/2] is a form of the potential energy. This parameter is the absorbed or released energy during a chemical reaction or change during a phase transition [37].The value of μ for Z is more negative (−4.79 eV) than that for E (−4.74 eV). The electrophilicity (ω) measures show the stabilization in energy when the system acquires an additional electronic charge from the environment. The electrophilicity index (ω = µ 2/2η) contains information about both electron transfer (chemical potential) and stability (hardness) and is a better descriptor of global chemical reactivity [38].The high values of electrophilicity index shows high capacity of the molecule to accept electrons. The electrophilicity index for E and Z is 6.35 and 6.07 eV, respectively. E has the high erelectrophilicity index, therefore the E form has a higher capacity for accept electrons. The dipole moment (µ D ) is a good measure for the asymmetric nature of a structure [29]. The size of the dipole moment depends on the composition and dimensionality of the 3D structure. As shown in Table 3, two structures have high values for the dipole moment, which reflects that there is no symmetry in the structures. The dipole moment for Z(7.854 Debye) is higher than for E(7.577 Debye).
Molecular Electrostatic Potential (MEP)
The molecular electrostatic potential (MEP) was calculated at the B3LYP/6-311+G* level. The MEP shows the electronic density and is useful for mapping sites for electrophilic attack and nucleophilic reactions as well as hydrogen bonding interactions [39]. Negative areas of the MEP (red color) are related to electrophilic reactivity and positive areas (blue color) to nucleophilic reactivity. Neutral regions are colored green, as shown in Fig. 7. According to the MEP maps presented in Fig. 7, there are two sites on E and Z for a possible electrophilic attack such as oxygen atoms of carbonyl groups of theisatin ring (higher color intensity) and the negative region is mainly focused on these atoms, therefore they are sites for electrophilic attraction. As the MEPs show, most regions in the two structures are neutral (green color).

Molecular electrostatic potential (MEP) maps for E and Z isomers, calculated using the B3LYP/6-311+G* level
Thermodynamic Analysis
The thermodynamic calculations were performed using DFT in the gas phase and E0 (sum of the electronic and the zero-point energies), enthalpy H (sum of the electronic and the thermal enthalpy), Gibbs free energy G (sum of the electronic and the thermal free energy), entropy S and Cv (constant volume molar heat capacity) were obtained (see Table 4) [40]. Thermodynamic analyses indicate that E0, H, and G for E and Zare negative values whereas the calculated S is positive that shows two molecules to be stable in the gas phase. Also we found that Z is more stable than E.
NBO Analysis
Natural bond orbital (NBO) analysis is an important method for studying intra- and intermolecular bonding and interactions between bonds [41, 42]. Electron donor orbital, acceptor orbital and the interacting stabilization energy resulting from the second-order micro disturbance theory [43] are reported in Table 5. The electron delocalization from filled NBOs (donors) to the empty NBOs (acceptors) describes a conjugative electron transfer process between them [44]. For each donor (i) and acceptor (j), the stabilization energy E (2) associated with the delocalization i→j is estimated. The resonance energy (E (2)) detected the quantity of participation of electrons in the resonance between atoms [23]. In E, LP(1)N1 orbital participates as donor and the anti-bonding BD*(2)C12–C13, BD*(2)C19–O3, BD*(1)C4–C5 and BD*(2)C4–C5 orbitals act as acceptor and their resonance energies (E(2)) is 37.00, 46.91, 3.14 and 7.50 kcal/mol, respectively. These values indicate large charge transfer from the LP(1)N1 to the anti-bonding orbital of BD*(2)C19–O3 [LP(1)N1→BD*(2)C19–O3] and BD*(2)C12–C13 [LP(1)N1→BD*(2)C12–C13]. Therefore there is a strong intramolecular hyperconjugative interaction between the nitrogen lone pair [LP(1)N1] with C19–O3 and C12–C13 antibonding orbitals. The LP(1)N1 orbital participates as donor and the anti-bonding BD*(1)C4–C5, BD*(2)C4–C5, BD*(2)C12–C13 and BD*(2)C19–O3 orbitals act as acceptors, and their resonance energies (E (2)) are 4.37, 0.57, 35.15 and 45.61 kcal/mol, respectively. The most important interaction energy related to the resonance of the molecule is electron donation from LP(1)N1 to the antibonding acceptor orbitals BD*(2)C12–C13 (35.15 kcal/mol) and BD*(2)C19–O3 (45.61 kcal/mol).
Conclusion
In summary, two isomers (E and Z) of ethyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate were prepared and characterized by FT-IR, 1HNMR, 13CNMR and X-ray crystallography. The electronic properties and geometric parameters have been analyzed with DFT calculations (B3LYP/6-311+G*). The theoretical results and the experimental data have been found to support each other. The HOMO–LUMO energy gap calculated at the B3LYP/6-311+G* level reveals the chemical activity and kinetic stability of the molecules. The NBO analysis confirms the hyperconjugation interaction. The thermodynamic calculations reveal that Z is more stable than E.
Notes
CSD refcodes for the 10 examples of 2-N-substituted ethyl 3-phenylprop-2-enoates: CEMBEB, EDOSUM, FETTAA, KITNIL, KITNOR, OWAGUP, OWAHAW, QIPZEW, YINGAE, OYOMIA.
References
Paira M, Banerjee B, Jana S, Mandal SK, Roy SC (2007) Tetrahedron Lett 48:3205–3207
Hoffmann HMR, Rabe J (1984) Helv Chim Acta 67:413–415
Alizadeh A, Rostamnia S, Zhu LG (2006) Tetrahedron 62:5641–5644
Hoffmann HMR, Rabe J (1985) J Org Chem 50:3849–3859
Lienafa L, Mongeand S, Robin JJ (2009) Eur Polym J 45:1845–1850
Baskaran D (2003) Prog Polym Sci 28:521–581
Feng L, Zhang Z, Wang F, Wang T, Yang S (2014) Fuel Processing Technol 118:42–48
Cramer CJ (2002) Essentials of computational chemistry: theories and models. Wiley, Chichester
Avci D, Atalay Y (2009) Struct Chem 20:185–201
Nazarski RB (2009) J Phys Org Chem 22:834–844
Shoaei SM, Kazemizadeh AR, Ramazani A (2011) Chin J Struct Chem 30:568–574
Hopfl H, Gomez B, Martinez-Palou R (2005) J Mex Chem Soc 49:307–311
Diffraction Oxford (2009) CrysAlis CCD and CrysAlis RED in KM4-CCD Software. Oxford Diffraction Ltd, Abingdon
Sheldrick GM (2008) Acta Crystallogr Sect A 64:112–122
Sheldrick GM (2015) Acta Crystallogr Sect C 71:3–8
Brandenburg K, Putz H (2012) Diamond–crystal and molecular structure visualization. Crystal Impact GbR, Bonn
Hariharan PC, Pople JA (1973) Theor Chim Acta 28:213–322
Hariharan PC, Pople JA (1974) Mol Phys 27:209–214
Kohn W, Becke AD, Parr RG (1996) J Phys Chem 100:12974–12980
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03, revision B03, Gaussian Inc., Pittsburgh
Frisch A, Nielson AB, Holder AJ (2000) GAUSSVIEW User Manual, Gaussian Inc., Pittsburgh
Seyed Katouli SA, Sheikhi M, Sheikh D, Tayyari SF (2013) Orient J Chem 29:1121–1128
Sheikhi M, Sheikh D (2014) Rev Roum Chim 59:761–767
Ramazani A, Noshiranzadeh N, Mahyari AT, Ślepokura K, Lis T, Ahankar H, Amini I (2007) Anal Sci 23:x195–x196
Mahyari AT, Shajari N, Kazemizadeh AR, Ślepokura K, Lis T, Ramazani A (2007) Z Naturforsch B 62b:829–834
Noshiranzadeh N, Ramazani A, Mahyari AT, Ślepokura K, Lis T (2008) Z Naturforsch B63b:65–70
Ramazani A, Farshadi A, Mahyari A, Ślepokura K, Lis T, Rouhani M (2011) J Chem Crystallogr 41:1376–1385
CSD, Groom CR, Allen FH (2014) Angew Chem Int Ed 53:662–671
Habibi D, Faraji AR, Sheikh D, Sheikhi M, Abedi S (2014) RSC Adv 4:47625–47636
Sheikhi M, Mahmoodi Hashemi M, Monajjemi M (2014) Orient J Chem 30:345–350
Sebastian S, Sundaraganesan N (2010) Spectrochim Acta A 75:941–952
Luque FJ, Lopez JM, Orozco M (2001) Theor Chem Acc 103:343–345
Soltani A, Ghari F, Dehno Khalaji A, Tazikeh Lemeski E, Fejfarova K, Dusek M, Shikhi M (2015) Spectrochim Acta A 139:271–277
Soltani AR, Baei MT, Mirarab M, Sheikhi M, Tazikeh Lemeski E (2014) J Phys Chem Solids 75:1099–1105
Pearson RG (2005) J Chem Sci 117:369–377
Das KGV, Panicker CY, Narayana B, Nayak PS, Sarojini KB, Al-Saadi AA (2015) Spectrochim Acta A 135:162–171
Koopmans TA (1993) Physica 1:104–113
Parr RJ, Szentpaly LV, Liu S (1999) J Am Chem Soc 121:1922–1924
Sheiri L, Sheikh D, Sheikhi M (2014) Rev Roum Chim 59:825–834
Shiri L, Sheikh D, Faraji AR, Sheikhi M, Seyed Katouli SA (2014) Lett Org Chem 11:18–28
Monajjemi M, Sheikhi M, Mahmodi Hashemi M, Molaamin F, Zhiani R (2012) Inter J Phys Sci 7:2010–2031
Weinhold F, Landis CR (2001) Natural bond orbitals and extensions of localized bonding concepts. Chem Educ Res Pract 2:91–104
Joshi BD, Tandon P, Jain S (2012) Himal Phys 3:44–49
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1998) NBO version 3.1,TCI. University of Wisconsin, Madison
Acknowledgements
This work is funded by the Grant NRF-2015-002423 of the National Research Foundation of Korea.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ramazani, A., Sheikhi, M., Ahankar, H. et al. Crystal Structure, Spectroscopic and DFT Studies on E and Z Isomers of Ethyl 2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-propenoate. J Chem Crystallogr 47, 198–207 (2017). https://doi.org/10.1007/s10870-017-0697-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-017-0697-8