
Abstract
The yeast mitochondrial ATP synthase is a rotary molecular machine primarily responsible for the production of energy used to drive cellular processes. The enzyme complex is composed of 17 different subunits grouped into a soluble F1 sector and a membrane-embedded F0 sector. The catalytic head of the F1 sector and the membrane integrated motor module in the F0 sector are connected by two stalks, the F1 central stalk and the F0 peripheral stalk. Proton translocation through the F0 motor module drives the rotation of the subunit 910-ring that generates torque which is transmitted to the calaytic head through the γ subunit of the central stalk. The rotation of the γ subunit causes changes in conformation of the catalytic head which leads to the synthesis of ATP. Biogenesis of the enzyme involves modular assembly of polypeptides of dual genetic origin, the nuclear and the mitochondrial genomes. Most of the yeast ATP synthase subunits are encoded by the genome of the nucleus, translated on cytosolic ribosomes and imported into mitochondria. In the mitochondria, the enzyme forms a dimer which contributes to the formation of cristae, a characteristic of mitochondrial morphology. Substantial progress has recently been made on the elucidation of detailed stucture, function and biogenesis of yeast mitochondrial ATP synthase. The recent availability of high-resolution structure of the complete monomeric form, as well as the atomic model for the dimeric F0 sector, has advanced the understanding of the enzyme complex. This review is intended to provide an overview of current understanding of the molecular structure, catalytic mechanism, subunit import into mitochondria, and the subunit assembly into the enzyme complex. This is important as the yeast mitochondrial ATP synthase may be used as a model for understanding the corresponding enzyme complexes from human and other eukaryotic cells in physiological and diseased states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Organization and topography of yeast mitochondrial ATP synthase
Adenosine triphosphate synthase (ATP synthase) also known as F1F0-ATPase, ATP phosphohydrolase, E.C.3.6.13, is a molecular motor that plays a central role in the production of energy used to drive cellular processes. As one of the most important enzymes, ATP synthase is responsible for generation of more than 90% of cellular ATP of eukaryotic cells under aerobic conditions (Kabaleeswaran et al. 2006; Srivastava et al. 2018). The ATP, also called “energy currency”, is a primary carrier of energy in the cells. The ATP synthase is generally found in the plasma membrane of bacteria, the inner mitochondrial membrane of eukaryotic cells and the thylakoid membrane of chloroplast. Mitochondrial ATP synthase catalyzes the formation of adenosine triphosphate (ATP) from adenosine diphosphate (ADP) and inorganic phosphate (Pi) at the expense of the transmembrane electrochemical proton gradient generated by the respiratory electron transport chain. The proton gradient needed to drive ATP synthesis is produced by other types of proton pumps powered by substrate oxidation. The reaction catalyzed by the ATP synthase is reversible. The enzyme can use the energy of ATP to drive protons uphill (into the mitochondrial intermembrane space, a region of higher H+ concentration) and form a transmembrane proton gradient which can be used to facilitate ion transport, substrate uptake, etc. (Tzagoloff and Myers 1986; Attardi and Schatz 1988; Guo et al. 2017). The enzymes is comprised of two functional and separable sectors, F1 and F0 sectors, both of which can function as molecular motors. When F1 hydrolyses ATP, the enzyme pumps protons in the opposite direction, and if F1 is detached from F0, the enzyme is only able to catalyse ATP hydrolysis (Junge et al. 1997).
Yeast provides a useful system for the analysis of structure, function, and biogenesis of mitochondrial ATP synthase. The well established molecular genetic techniques have permitted the manipulation the genes coding for subunits of ATP synthase in order to elucidate stoichiometry, structure and function of the enzyme complex. The yeast system is also effective for identification of ATP synthase subunits (Devenish et al. 2000). Advances in X-ray crystallography and cryo-electron microscopy (cryo-EM) techniques have provided better insights on the structure, function and biogenesis of the enzyme, as well its roles in the formation of cristae in the inner mitochondrial membrane (Hahn et al. 2016).
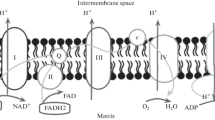
ATP synthase from different sources shows a similar basic structure. The yeast mitochondrial ATP synthase is an assembly of 30 subunits of 17 kinds (Table 1) with a molecular weight of about 600 kDa (Xu et al. 2015). The subunits of the enzyme complex are divided into two separable sectors, F1 and F0 sectors (Fig. 1). The water soluble F1 sector is located in the matrix and comprised by subunits α, β, γ, δ and ε with stoichiometry of 3:3:1:1:1. It forms a spherical globular structure and contains the catalytic sites. All of the subunits of the F1 sector are encoded by nuclear genes. The F0 sector is composed of subunits b, OSCP (oligomycin sensitivity conferring protein), d, e, f, g, h, i/j, k which are encoded by nuclear genes, and subunits 6, 8, and 9, which are encoded by mitochondrial genes. The F0 sector is embedded to the inner membrane of mitochondria (Cox et al. 1992; Devenish et al. 2000).

Schematic representation of monomeric form of yeast mitochondrial ATP synthase (Adapted and modified from Devenish et al. 2000). The ATP synthase consists of the matrix-exposed F1 sector and the inner membrane-bound F0 sector. In the membrane-extrinsic F1 sector, subunits α and β are shown alternately arranged with subunit γ lying in the centre (Srivastava et al. 2018). Together with subunits δ and ε, subunit γ is shown to comprise the central stalk. Subunits δ and ε are shown to be bound at the foot of the subunit γ (Kabaleeswaran et al. 2006). The catalytic head of the F1 sector and the membrane-integrated rotor module of the F0 sector are linked by two stalks, the F1 central stalk and the F0 peripheral stalk The peripheral (stator) stalk is depicted as being comprised by subunits b, d, OSCP, and h (Bateson et al. 1999; Kabaleeswaran et al. 2006). In the membrane-embedded F0 sector, 10 identical monomers of subunit 9 (more commonly known as subunit c, using bacterial nomenclature) are shown arranged in a ring-like structure (Guo et al. 2017). Subunits 8, subunit 6 and other subunits with transmembrane topology are shown. Subunit 6 (subunit a), together with the subunit 910-ring, forms the proton-conducting channel of the F0 region. Transport of protons across the F0 rotor module results in rotation of the subunit 910-ring. The torque of the c-ring is transmitted to the catalytic head of the F1 sector through the γ-subunit of the central stalk. The three heterodimers subunits α and β in the F1 sector that form the reactive centers undergo conformational changes upon rotation of subunit γ, leading to synthesis of ATP. The OSCP on the top of F1 head acts as a flexible hinge between F1 and the peripheral stalk, preventing idle rotation of the catalytic head with F0 (Kühlbrandt 2019). Supernumerary subunits part of the F0 region contribute to the formation of dimers of the ATP synthase (Song et al. 2018)
The activity of the yeast mitochondrial ATP synthase is regulated in order to overcome wasteful hydrolysis of ATP when the oxygen supply to the electron transport chain is limited (Devenish et al. 2008). Three proteins, INH, STF1 and STF2 belong to an inhibitor complex. The protein INH is a natural inhibitor protein that plays role in the direct regulation of the enzymatic activity. The inhibitory action of INH protein is enhanced by two proteins, STF1 and STF2 which function as stabilizing factors (Dienhart et al. 2002). Studies in mammalian ATP synthase indicated that the inhibitor of the enzyme is active in its dimeric form under acidic conditions such as during ischemia/hypoxia when the matrix pH drops below 7, or when the H+ electrochemical gradient is lost (Faccenda and Campanella 2012; Kühlbrandt 2019). The yeast INH protein is a homologue of the mammalian IF1 protein. The mammalian IF1 protein can inhibit the activity of the yeast mitochondrial ATP synthase. In contrast, yeast INH protein is unable to inhibit the mammalian enzymes because the INH protein is stabilized by the STF1 and STF2 proteins which have no homologues in the mammalian mitochondria (Faccenda and Campanella 2012).
There are in total six nucleotide binding sites in the F1 sector, three of which are catalytically active, and the other three have no direct involvement in the enzyme catalytic function (Senior et al. 1995). F1 sector has a central stalk (rotor) comprised of three subunits, the subunits γ, δ, and ε. At one end the rotor is within the core of F1 and, at the other end, is connected to the F0 sector. Each of the three catalytic sites in F1 cycles between three different states and shows asymetrical position relative to the subunit γ (Srivastava et al. 2018). The N- and C-termini of subunit γ form α-helix secondary structure. These regions, together with a foot composed of an α/β bundle, are involved in the formation of coiled-coil part of the subunit which is located within the α/β core of the enzyme complex. The subunit δ is bound to the foot of subunit γ. It consists of a β-sandwich composed of two 5-stranded β-sheets and two α-helixes. As a component of the central stalk, subunit ε is also bound at the foot of subunit γ. It has a loop region formed by two α-helixes with extended parts of polypeptide at the N- and C-termini. When the yeast subunit ε is compared to that of the bovine, it has 10 extra amino acids at the C-terminus. This region folds back and is situated under the central core of the central stalk (Kabaleeswaran et al. 2006).
The F0 sector of yeast mitochondrial ATP synthase is much more complex than that of bacteria. In yeast, the F0 sector is composed of at least 12 polypeptides. Only 4 of them, subunits b, OSCP, 6, and 9, have homologues in the bacterial enzyme, termed b, δ, a, and c subunits, respectively (Table 1). The rest, that includes subunits 8, e, f, g, h, i/j, and k, have no homologues in the bacterial complex. This implies that the bacterial enzyme can function well in the absence of these subunits. This phenomenon has raised intriguing questions on the roles of these additional subunits in the yeast ATP synthase. Studies in yeast therefore have also been directed to elucidation of structures and roles of these subunits in the enzyme complex (Devenish et al. 2000). Subunit 8 is an intrinsic membrane protein of 48 amino acids essential for the enzyme assembly and function (Roucou et al. 1999; Artika 2007). It has a single transmembrane domain with its N-terminus located in the intermembrane space and its C-terminus is located in the matrix (Stephens et al. 2000). It is obligatory for subunit 8 to maintain a transmembrane topology for providing proper functioning as part of the stator stalk (Artika 2009). It has been suggested that subunit 8 is in close proximity to subunits b, d, and f in the matrix and to subunits b, f, and 6 in the intermembrane space (Stephens et al. 2003). High resolution structural data showed that subunit 8 is almost entirely adopts an α-helix structure. The N-terminus part of the helix was shown to be embedded in the membrane in contact with subunit 6, and the C-terminus 14 residues was shown to be part of the base of the stator stalk (Guo et al. 2017). Evolutionarily, the yeast subunit 8 is thought to be derived from one of the two copies of the bacterial subunit b based on the N-terminal MPQL motif (Hahn et al. 2016).
Subunit f is a protein of 95 amino acid residues essential for mitochondrial respiration (Spannagel et al. 1997) that has a soluble N-terminus of about 50 amino acid residues and a single transmembrane α helix (Guo et al. 2017). Subunit h is a hydrophilic polypeptide of 92 amino acid residues which is essential for ATP synthase activity (Arselin et al. 1996). Subunit i/j is an amphiphilic polypeptide of 59 amino acid residues important for the activity of the enzyme complex (Vaillier et al. 1999; Arnold et al. 1999). Subunits e, g and k are dispensable for formation of the enzymatically-active ATP synthase complex (Xu et al. 2015). All of these additional subunits are now assigned to being components of the stator stalk. In addition to their roles as component of the stator stalk, most of the additional subunits have also been shown to be involved in the formation of the enzyme dimer (Guo et al. 2017).
The term F0 refers to a factor that confers sensitivity of the F1 ATP synthase to the antibiotic, oligomycin. Although mitochondrial ATP synthase has been studied for more than 50 years, the oligomycin binding site on the F0 sector has been difficult to find. It is now known that that oligomycin binds to subunit 9 and blocks proton passage through the F0 sector. High-resolution structures of yeast mitochondrial ATP synthase have shown that oligomycin forms an hydrogen bond with the carboxyl-side-chain of Glu59 of the subunit 9 (Symersky et al. 2012a). The core of the F0 sector is comprised of subunit 6 (commonly known as subunit a) and subunit 9 (more commonly known as the c subunit). In the membrane-embeded F0 sector, subunit 9 proteins assemble to form a ring-like structure which functions as a rotor. A striking difference between F1F0-ATP synthases from various organisms, is the size of their subunit 9-ring rotors. The number of the subunits 9 forming the ring ranges from 8 in mammalian mitochondrial ATP synthase, to 14 in chloroplast, to 12 in E. coli, to 15 in cyanobacteria, and to 17 in Burkholderia pseudimallei (Kühlbrandt 2019). High resolution structural data has clearly shown that in yeast, the ring-like stucture of the F0 sector is composed of 10 copies of subunit 9 (commonly termed c10-ring, using bacterial nomenclature). The subunit 6 is located adjacent to the subunit 910-ring and the horizontal helices of subunit 6 wrap around the subunit 910-ring rotor. Together with the subunit 910-ring, the subunit 6 participates in the formation of the proton conduction pathway from mitochondrial intermembrane space side to the protonation sites in the subunit 910-ring and from these sites to the matrix. They form 2 aqueous half-channels on either side of the inner mitochondrial membrane (Xu et al. 2015; Hahn et al. 2016; Srivastava et al. 2018).
Compared to the relatively slim bacterial and chloroplast stator stalks, which are thin as they are composed of only 2 subunits b (subunits b and b’ in the case of chloroplast) and one subunit δ (homologue of eukaryotic subunit OSCP) at the top of F1, the stator stalk of yeast mitochondrial ATP synthase is more bulky (Kühlbrandt 2019). It is composed of subunits 6, OSCP, b, d, h, f, i/j, and 8. Consistent with studies in other organisms (Devenish et al. 2008), high resolution cryo-EM structures showed that the yeast subunit OSCP which is on the top of the F1 head, interacts with the N-terminus of the three α-subunits to stabilize its binding to the top of F1 (Srivastava et al. 2018) as illustrated in Fig. 1. This is consistent with previous data showing that the N-terminal region of yeast OSCP interacts with the N-terminal regions of α-subunits on top F1 sector, distal from the F0 membrane sector. The OSCP subunit extends along the the surface of F1 down towards F0, where it interacts with the C- terminus of the b subunit (Rubinstein and Walker 2002). The OSCP functions as a flexible hinge connecting the F1 head and the peripheral stalk preventing idle rotation of the catalytic head with F0 (Kühlbrandt 2019). The yeast subunt b (also known as subunit 4) was shown to extend from the membrane to the top of the F1 subcomplex (Davies et al. 2012). The peripheral stalk of the yeast Saccharomyces cerevisiae mitochondrial ATP synthase was shown to have a similar composition to that of the bovine enzyme. Studies in bovine mitochondrial ATP synthase indicated that a heterodimer is formed by subunits OSCP and b interacting with subunits d and h (or F6) (Rubinstein et al. 2005). Although the yeast subunit h is a weak homologue of the bovine subunit F6, the function of yeast subunit h can be replaced by the bovine subunit F6. The yeast subunit h was shown to be a component of the peripheral stalk with its C-terminal region lying in a region of the peripheral stalk close to the F0 membrane sector (Rubinstein et al. 2005). The yeast subunit d is predominantly hydrophilic protein composed of 173 amino acids long homologous to the bovine subunit d (Norais et al. 1991). Subunit d is found only in yeast and other mitochondrial ATP synthases, not in the bacterial and chloroplast enzymes. It is an essential subunit of the mitochondrial ATP synthases. In the peripheral stalk subunit d interacts with all other components of the stalk, subunits OSCP, b, and h (Devenish et al. 2008). The stator stalk plays a role in bridging F0 with F1, along the noncatalytic site, which involves contact between subunits α and d. Below the F1 head, the peripheral stalk subunits b and d bend toward the central stalk (Hahn et al. 2016; Srivastava et al. 2018). Recent high resolution cryo-electron microscopic structural data showed that subunits 8, f and b contribute to the base of the stator stalk, with subunit d acting as a clip (Guo et al. 2017).
Catalytic mechanism of ATP synthase
According to the chemiosmotic hypothesis postulated by the biochemist Peter Mitchell in 1961, electron transport and ATP synthesis are coupled by a proton gradient across the inner mitochondrial membrane (Mitchell 1961). In this hypothesis, the transfer of electrons through the respiratory chain leads to the pumping of protons from the matrix to the mitochondrial intermembrane space. The proton (H+) concentration is higher on the intermembrane space side, and an electric potential with this side positive is generated. Mitchell postulated that this proton-motive force drives the synthesis of ATP by the ATP synthase as protons flow back into the matrix through a proton pore associated with the enzyme complex. Therefore, the primary energy-conserving event according to this model is the movement of protons across the inner mitochondrial membrane. This highly innovative hypothesis that oxidation and phosphorylation are coupled by a proton gradient is now supported by a wealth of current data (Srivastava et al. 2018).
How energy derived from a proton motive force is used to synthesize ATP is further explained by the concept of the binding change mechanism proposed by Paul Delos Boyer (Boyer 1975). According to this mechanism, the structure of the three catalytic sites is always different, each passing through a cycle of “open” (O), “loose” (L) and “tight” (T) states. The open site has very low affinity to ligands and is also catalytically inactive. While the tight site with tightly bound ligands is catalytically active. The translocation of proton triggers conformational change which converts the T-site with bound ATP to an O-site, releasing the bound nucleotide. At the same time, an L-site with loosely bound ADP and phosphate is converted to a T-site, where the substrates are tightly bound and ATP is formed (Cross 1992; Abraham et al. 1994). A very simplified version of the catalytic mechanism of ATP synthase is depicted in Fig. 2. The presence of three catalytic sites in different configurations has previously been shown in crystal structure of bovine F1 which was grown in the presence of Mg2+. It was observed that one catalytic site was filled with ADP, another with AMP-PNP (a non-hydrolysable ATP analogue) and the third site was empty (Abraham et al. 1994). Similarly, the high resolution structures of yeast F1 sector shows asymmetric conformational features which is nearly identical to that observed in the crystal structures of the bovine F1, revealing the three distinct states of the catalytic sites: O, L, and T (Kabaleeswaran et al. 2006; Srivastava et al. 2018).

A very simplified version of the binding-change-mechanism for ATP synthesis by F1F0-ATP synthase. The enzyme complex is viewed from the top of F1. The central arrow represents the γ subunit. In step 1, rotation of asymmetric γ subunit forces conformational changes for substrates and product at the catalytic sites. T, L, and O stand for tight, loose, and open conformation respectively and refer to deceasing affinities of catalytic sites to ligand. Each of the three coloured area represents a pair of α and β subunits, where the catalytic sites are formed at the interface of the two subunits. In this illustration, the αβ pairs remain stationary. In step 2, ATP forms spontaneously from tightly bound ADP and Pi. The open site is ready to bind fresh substrates and so on. Adapted from Cross (1992) and Cross and Duncan (1996)
According to the binding-change-model for ATP synthesis, the three catalytic sites of the enzyme complex work cooperatively by binding ADP and Pi in sequence before undergo a conformational change that leads to spontaneous formation of ATP from the tightly bound ADP and Pi. The newly formed ATP is tightly bound. The sites then undergo conformational change again in order to release the ATP. These changes in the binding affinities of reactants and product are induced by rotational catalysis powered by the rotating central stalk of the enzyme, which is in turn driven by the protons flow across the mitochondrial membrane. According to this mechanism, ATP cannot be released from one site unless ADP and Pi were available to bind to another catalytic site. Similarly, when the reaction ran in the reverse direction, Pi could not be released from one catalytic site unless ATP was available to bind at another site (Boyer 1989; Kresge et al. 2006). The rotary movement of subunit γ during rotational catalysis of ATP synthase has been directly visualized by Noji and colleagues using a single molecule of bacterial F1 (Noji et al. 1997). In these experiments recombinant, F1 from a thermophilic bacterium was immobilised head-down via engineered hexahistidine tags to a glass plate coated with horseradish peroxidase conjugated with Ni2+-nitrilotriacetic acid (Ni-NTA). A flourescent actin filament was attached through streptavidin to the γ subunit as a marker to allow observation of its motion directly (Fig. 3). It was found that in the presence of ATP, the filament rotated in an anticlockwise direction when viewed from the membrane side (Noji et al. 1997).

Diagramatic representation of a system used for observation of the γ-subunit rotation in the α3β3γ subcomplex. The α3β3 γ subcomplex with ten histidine (his tag) linked to the N-terminal of each β-subunit was fixed on a glass plate. The glass plate was coated with horseradish peroxidase conjugated with Ni2+-nitrilotriacetic acid (Ni-NTA), which has a high affinity for the His tag and thus bind the subcomplex through the three β-subunits, with the F0 side (membrane side) away from the glass. As a strategy to visualise the rotation, γ-Ser107, which is presumably in the stalk region of the γ-subunit was replaced with cysteine, and α-Cys193, the only cysteine in the wildtype α3β3γ subcomplex was replaced by serine. The introduced cysteine was biotinylated. A fluorescently labelled biotinylated actin filament was attached to the γ-subunit through streptavidin, which has four binding sites for biotin, Therefore, the actin filaments are attached to the biotinylated α3β3γ subcomplex which are fixed to the glass surface through His tags. The rotation of actin filament was observed by epiflourescence microscopy (not shown in the diagram) and the images were taken with a camera attached to the image intensifier (Noji et al. 1997). Figure is adapted from Noji et al. (1997)
The yeast mitochondrial ATP synthase utilises the potential energy from transmembrane electrochemical proton gradient generated by respiration for synthesis of ATP in an endergonic reaction. Protons have the potential to flow from high to low concentration thereby generate torque in the F0 rotor, the subunit 910 ring (more comonly termed c10 ring). To transclocate through the membrane, protons are believed to bind sequentially to subunits of the ring rotating within the membrane. This F0 turbine is attached to central stalk that enable mechanical energy to be transferred to F1 and ushes the catalytic subunits fo F1 into different conformations to provide the correct chemical environment for ATP synthesis from its building blocks of ADP and inorganic phosphate, Pi. In the reverse reaction, the ATP synthase provide different chemical environment for binding of ATP, followed by hydrolysis, and finally release of product ADP and Pi (Stewart et al. 2014).
How protons travel through the F0 region was recently clarified by the structural data of the F0 complex from yeast (Guo et al. 2017; Srivastava et al. 2018). Two pores corresponding to two half-channels in the F0 sector are required for proton translocation. The cavity observed on the inter membane space side forms the inter membrane space half-channel and the opening on the matrix side between subunit 6 and the subunit 910-ring forms the matrix side half-channel (Guo et al. 2017). The inter membrane-half channel is formed by residues in the subunits f, b, and 6. The matrix side-half channel at the interface of subunits 6 and 9 is formed by a number of residues that create a hydrophilic cavity that extends from a subunit 9-Glu59 to the surface of the membrane space (Srivastava et al. 2018). The passage of protons from inter membrane space to the matrix drives rotation of the subunit 910-ring in 36o steps, and in turn, rotates the central rotor within the F1 in 120o steps. The rotation of the central rotor causes conformational changes in the catalytic sites that provides the energy required for phosphorylation of ADP, as proposed by the binding-change-hypothesis of Paul D Boyer. Therefore, the protonation and deprotonation events at the interface between subunits 6 and subunit 910-ring couple the translocation of protons to the rotation of subunit 910-ring and thus drives ATP synthesis. As the central rotor couples the rotation within the F1 core, the futile rotation of F1 is prevented by the stator stalk connecting F1 with F0 (Srivastava et al. 2018).
The yeast subunit 9 is a polypeptide of 76 amino acid residues with conserved Glu59 which has been postulated to play roles as the proton acceptor and donor at the interface of the subunit 910-ring with subunit 6. The Glu59 is reponsible for the net proton translocation during the catalytic cycle. It was shown in the crystal structures of subunit 910 rings that the side chain of Glu59 provides a proton (H+)-binding site at each of the interface between subunits 9. This side chain is in closed conformation when in the membrane and in open conformation only when at the interface between subunits 9 and 6. It is hypothesized that the closed conformation represents the protonated state and the open conformation represents the state in which protonation and deprotonation is occuring (Symersky et al. 2012b; Srivastava et al. 2018). Subunit 6 is a polypeptide of 249 amino acid residues with strictly conserved Arg176. This residue has been postulated to play roles in coordinating deprotonation and protonation events, preventing uncoupled proton leakage and acting as a positive pole to attract the negatively charged Glu59. During ATP synthesis, proton transit is from its cytosolic side of the mitochondrial membrane to protonate the subunit 9-Glu59, prior to the protons being released into the matrix space (Symersky et al. 2012a, b; Guo et al. 2017; Srivastava et al. 2018).
Biogenesis of yeast mitochondrial ATP synthase
The presence of organelles defines the existence of eucaryotic cells. Mitochondria are double-membrane-bounded organelles which principally function in the processes related to cellular energy production such as respiration and oxidative phosphorylation. As organelles bearing specialized functions, they generally contain distinct sets of proteins and other molecules. In yeast, there are more than 1000 different proteins functioning in the mitochondria. Mitochondria possess their own genome, ribosomes, and all the enzymatic machinery required for mitochondrial gene expression. However, most of the mitochondrial proteins are encoded in the cell nucleus, translated as precursors on cytosolic ribosomes and imported into mitochondria. Only eight of the yeast mitochondrial proteins are encoded by the mitochondrial genome and synthesized within the organelle (Attardi and Schatz 1988; Fox 2012).
The mitochondrial ATP synthase is a major constituent of mitochondria comprising about a quarter of the membrane protein. The biogenesis of mitochondrial ATP synthase complex involves the two separate genomes, the nuclear and the mitochondrial genomes. The two genomes collaborate in the synthesis and assembly of ATP synthase subunits and in the regulation of mitochondrial energy production. Most of the mitochondrial ATP synthase subunits are encoded by nuclear genes, translated in the cytosol and then targeted to specific mitochondrial sites. In yeast, three subunits of mitochondrial ATP synthase are encoded by mitochondrial genes and translated on mitochondrial ribosomes. They are subunits 6, 8, and 9, all of them are belong to the F0 sector. The remainder subunits are specified by nuclear genes, translated in the cytosol and then imported into mitochondria. All of the yeast F1 subunits are encoded by nuclear genes (Sebald and Hoppe 1981; Attardi and Schatz 1988; Nagley 1988). In mammalian mitochondrial ATP synthase, the subunit 9 polypeptide is encoded by three nuclear genes, translated on the cytosolic ribosomes and imported into the mitochondria (He et al. 2018).
In general, protein import into mitochondria is initiated by the recognition and binding of a precursor protein by specific import components in the cytosol, located at the cytosolic surface of the outer mitochondrial membrane, followed by the insertion of the preproteins into the outer membrane import sites (Hauckle and Lithgow 1997). Proteins to be imported into mitochondria are generally synthesized as a precursor containing signal peptides which play roles in targeting the proteins to mitochondria. Several protein import pathways have been indicated, each of which has a different type of targeting signal. Most of the matrix proteins and many inner-membrane proteins are imported using the so-called presequence pathway. They are typically synthesized with N-terminal presequences that function as targeting signals. The size of the presequence varies, usually from 19 to 50 amino acid residues. The presequences (signal peptides) usually form an amphipathic α-helix that harbours a positively charged face and a hydrophobic face. The elements of the amphipathic helix are specifically recognized by the mitochondrial membrane receptors and other import machineries during preprotein translocation (Hartl et al. 1989; Wiedemann and Pfanner 2017).
Import into mitochondria requires the precursor proteins to be in a competent state for import. Molecular chaperones such as Hsp (heat shock protein) 70 and ATP- dependent defolding enzymes have been suggested to take part in preventing the aggregation of precursors by protecting their hydrophobic regions, guiding preproteins to their specific receptors and maintaining them in a loosely-folded configuration. Hsp70 molecular chaperones are present in both cytosolic and mitochondrial compartments. Hsp70 directly interacts with translocating preproteins. It contains a peptide-binding domain that interacts with a short segment unfolded preprotein in its ATP-bound state. Cycles of interaction with precursor proteins is an important aspect of its function in protein translocation and folding. ATP-dependent release from the cytoplasmic Hsp70, leads to the translocation of the presequence-carrying preproteins by the translocases of the outer and inner mitochondrial membranes together with the presequence translocase-associated motor into the matrix, where the preproteins interact with mitochondrial Hsp70 and the presequences are cleaved off by the mitochondrial processing peptidase. Following ATP-dependent release from the mitochondrial Hsp70, the majority of the proteins imported into the matrix require the assistance of Hsp60 for refolding and then assembly into oligomeric complexes (Cheng et al. 1989; Ting et al. 2014; Craig 2018) (Fig. 4).

Protein import into mitochondrial matrix. After the initial recognition of signal peptide by the mitochondrial membrane receptor and then insertion of the signal peptide and of the adjacent portions of the polypeptide chain, the unfolded chain slides in a channel that spans both membranes formed by the translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM). The import process requires hsp70 on both sides of the mitochondrial double membrane. Bound cytosolic hsp70 is released from the protein in a step that depends on ATP hydrolysis. Concomitantly, mitochondrial Hsp70 binds to regions of the polypeptide chain as they become exposed in the matrix, thereby pulling the protein into the interior of the mitochondrion. Following ATP-dependent release from the mitochondrial Hsp70, the proteins imported into the matrix require the assistance of Hsp60 for refolding and then assembly into functional oligomeric complexes
Yeast has been a useful model organism for identification of components and mechanisms that drive translocation and sorting of protein imported into mitochondria. Studies in yeast elucidated the components of translocases and provide information on their functions (Sokol et al. 2014). The translocase of the outer membrane (TOM) of mitochondria contains several proteins. Three proteins, Tom20, Tom22, and Tom70 function together as the receptor for precursor proteins. Tom70 is loosely attached to the TOM complex and is important for the import of noncleavable hydrophobic precursors. Presequences are recognized by Tom20 and Tom22 receptors and are directed into the translocation pore formed by the β-barrel protein Tom40. Tom40 guides the translocation of preproteins through the outer membrane. The interior of pore contains hydrophilic and hydrophobic regions that form distinct import paths for hydrophilic and hydrophobic precursors. In addition, there are three small proteins, Tom5, Tom6, and Tom7, that are not essential for the TOM functions but have been suggested to play roles in assembly and stability of the TOM complex. The presequence of the translocated protein is then handed over to the translocase of inner membrane (TIM), Tim23 complex. The yeast Tim23 complex is heteroterameric, being composed of two copies of Tim23 and two copies of Tim17. These two integral membrane proteins constitute the translocation channel. Tim23 forms a protein-conducting pore, whereas Tim17 has been suggested to play roles in regulating the Tim23 channel. This complex together with the import motor completes protein translocation into the matrix. Tim23 associates with Tim44 which serves as a scaffold for association with the motor components. The mitochondrial import process is believed to take place in the site of close contact between the outer and inner mitochondrial membranes (Fig. 5) (Sokol et al. 2014; Ting et al. 2014; Backes et al. 2018).

The protein import machinery of yeast mitochondria. Mitochondrial precursor proteins (preproteins) are synthesized with signal peptides that direct the proteins to mitochondria and into the correct location in the mitochondrial compartment. The preproteins synthesized in the cytosol and are imported by the translocase of the outer mitochondrial membrane (TOM) complex. The preproteins are then transferred from TOM to the translocase of the inner membrane (TIM23) complex. The proteins either are inserted into the inner membrane (IM) or are translocated into the matrix with the help of the presequence translocase-associated motor. The presequences are typically cleaved off by the mitochondrial processing peptidase. Adapted and modified from Wiedemann and Pfanner (2017). OM = outer mitochondrial membrane, IM = inner mitochondrial membrane, 70 = Tom70, 40 = Tom40, 22 = Tom22, 20 = Tom20, 7 = Tom7, 6 = Tom6, 5 = Tom5, 44 = Tim44, 23 = Tim23, 17 = Tim 17
The process of mitochondrial ATP synthase assembly is best characterized in yeast. It is a complicated process involving coordinated expression of nuclear and mitochondrial genomes. The enzyme complex is assembled from separate modules (Fig. 6). ATP synthase is therefore composed of different structural and functional units which jointly couple ATP synthesis or hydrolysis to proton translocation across the inner membrane. It has been suggested that the assembly of the F1 sector is independent of assembly of membrane integrated F0 sector. The assembly of F1 sector is promoted by the assembly factors Atp11 and Atp12. These chaperones bind to the newly imported subunits β and α, respectively. In the absence of these factors, subunits α and β aggregate and formation of active ATP synthase is blocked. The association of the central stalk leads to the release of Atp11 and Atp12 and the formation of the functional F1 sector (Rak et al. 2011; Song et al. 2018). Yeast Saccharomyces cerevisae assembly factors Atp11 and Atp12 have been shown to be the orthologs of the human ATPAF1 and ATPAF2 respectively which participate in the assembly of the human F1 sector (He et al. 2018).

The assembly pathway of yeast mitochondrial ATP synthase. The yeast mitochondrial ATP synthase is an assembly of 30 subunits of 17 kinds. The diagram shows the modular assembly of the yeast mitochondrial ATP synthase. The enzyme complex is composed of at least three different modules, F1, subunit 910-ring and subunits 8/6/stator subcomplex. The chaperones Atp12 and Atp11 promote formation of F1 domain. The newly translated subunit 9 oligomerizes into the ring before interacting with the F1 ATPase. Subunits 6 and 8 are associate with a peripheral stalk-F1 intermediate, and then followed by addition of the subunit 910-ring. Adapted from Rak et al. (2011) and Song et al. (2018)
Several assembly steps of the yeast mitochondrial ATP synthase have been proposed. Subunits 6 and 8 are thought to associate first with a peripheral stalk-F1 intermediate, followed by addition of the subunit 910-ring. In yeast, a number of assembly factors for formation of the membrane-bound F0 sector have been identified. The two-domain protein Atp25 stimulates synthesis and assembly of the subunit 9 into the subunit 9-ring. The protease Atp23 processes membrane-inserted yeast Atp6. In addition to its proteolytic activity, Atp23, along with the Atp10 chaperone, stabilizes and assembles yeast Atp6. The inner membrane assembly complex (INAC) binds to the newly assembled subunit 910-ring. It is thought that the INAC also binds to an assembly intermediate composed of the F1 domain, the peripheral stalk, subunit 6 and subunit 8, and the assembly factors Atp10 and Atp23. The INAC has a role in preventing any premature interaction (Song et al. 2018). The INAC is composed of Ina17 (17 kDa) and Ina22 (22 kDa) polypeptides. So far, no human orthologs are identified for these two proteins (He et al. 2018). An assembly intermediate composed of subunit 6, subunit 8 and stator subunits has also been identified. This assembly intermediate is stabilized by Atp10 (Rak et al. 2011).
One of the important steps in the assembly of yeast mitochondrial ATP synthase is the formation of the proton-conducting channel composed of subunit 6 and the ring of 10 identical subunits 9 as a rotor component of the Fo sector. The yeast subunit 9 is a proteolipid synthesized on mitochondrial ribosomes that are attached to the matrix side of the inner mitochondrial membrane. Its hydrophobic character leads to the suggestion that the subunit 9 may be inserted into the inner membrane co-translationally. The newly translated subunit 9 oligomerizes into the ring before interacting with the F1 component of ATP synthase. The F1-subunit 9 ring assembles with another module consisting of subunit 6, subunit 8, and all the peripheral stalk subunits except OSCP. In yeast, it was found that the assembly of the rotor component of the mitochondrial ATP synthase is enhanced when subunit 9 protein is in contact with subunit 6 of mitochondrial cytochrome c oxidase (Cox6) (Su et al. 2014). The next step is the assembly of the F1 central stalk with the F0 subunit 910-ring rotor, which mainly by electrostatic interaction (Dautant et al. 2010) (Fig. 6). It is interesting to note that several maturation steps in the assembly of the yeast mitochondrial ATP synthase complex differ from that of the human form. In yeast, subunits 6 and 8 associate first with a peripheral-F1 intermediate, followed by integration of the subunit 910-ring. In humans, subunits 6 and 8 combine to an enzyme intermediate form containing the F1 sector, peripheral stalk, and subunit 98-ring (c8-ring) (Song et al. 2018). This may be related to the fact that the yeast subunit 9 is mitochondrially encoded and translated in the mitochondrial matrix, while the human subunit 9 is nuclearly encoded and imported into mitochondria (He et al. 2018).
In general, the ATP synthase from different sources has a similar basic structure. However, the yeast mitochondrial ATP synthase differs from the bacterial and chloroplast ATP synthase, because the mitochondrial enzyme exists as a dimer. The dimer has a V-shaped structure of twofold symmetry, with an angle of 86° between its monomers. The monomers have been shown to interact within the membrane at the base of the peripheral stalks (Davies et al. 2012). The involvement of subunits e, g, k, i/j, b, and 6 in the dimeric formation of the enzyme complex has been suggested. Subunits e and g are thought to be responsible for inducing strong membrane curvature (Hahn et al. 2016). Subunits k and i/j, the two small subunits of the F0 sector, have been shown to be involved in the stepwise assembly of enzyme dimers (Wagner et al. 2010). Recent high-resolution cryo-electron microscopic structure of the dimeric F0 region of yeast ATP synthase has revealed that the subunits 6 and i/j form the contact sites between two ATP synthase monomers, being supported by interaction between subunits e and k. High resolution structural data also showed that the subunits e and g and the N-terminal transmembrane helix of subunit b are located at the interface between the two monomers in the dimer configuration. The interaction between these subunits is the reason for the unique V shape of the complex with the peripheral stalks project away from each other (Song et al. 2018). Subunits k and e are also thought to contribute to the final monomer-monomer interaction. Subunits 6, together with subunits i/j, k, and e has been reported to play roles in holding the yeast dimer together. The dimeric structure of the ATP synthase is also present in both bovine and human mitochondria. In the yeast dimeric F0 domain, subunit k has been proposed to associate with subunit 6 and is distant from the monomer-monomer interface. Based on its location in which it is suited to make contact between dimers in the inner mitochondrial membrane, the yeast subunit k has been proposed to be the ortholog of the human DAPIT protein. In humans, DAPIT is the most peripheral mitochondrial ATP synthase subunit and is suggested to function in the formation of links between ATP synthase dimers to generate the rows of dimeric complex to be found along the cristae edge (He et al. 2018). The yeast subunit i/j is thought to confer stability on the yeast ATP synthase dimer (Guo et al. 2017). Recent findings concluded that the yeast subunit i/j is a functional ortholog of the human 6.8 kDa proteolipid based on the sequences of the two proteins and the positions of their transmembrane α-helices. The human 6.8 kDa proteolipid plays roles in stabilizing the assembly process of subunits 6 and 8 to generate an active enzyme complex coupled to ATP formation (He et al. 2018).
In the inner membranes of yeast mitochondria, the ATP synthase units are organized with precision. The ATP synthase dimers are arranged into extending front-to-back rows located in the cristae region of the membrane especially along the sharply curved edges. The dimerization of ATP synthase units is postulated to play critical roles in the normal development of cristae morphology so as to maximize the surface area of inner mitochondrial membrane and thus maximize energy (ATP) production in the mitochondria. Quantitative molecular-simulation analysis suggested that a row-like organization of the ATP synthase dimers forms spontaneously, driven by attraction force arising from the release of an overall membrane elastic strain arising from the V-like structure of the dimers. Following their formation, the dimer-row-organization is thought to be a direct development of membrane folds and invaginations that eventually form the cristae. ATP synthase therefore is not only important for cellular bioenergetics, but also contributes to the formation of mitochondrial morphology (Anselmi et al. 2018).
Conclusion
Yeast has provided an attractive system for a deep understanding of the structure, function, and biogenesis of the mitochondrial ATP synthase complex. The established yeast genetic manipulation system has permitted the manipulation of genes encoding for ATP synthase subunits to enable elucidation of their stoichiometry, structure and topology, function, mehanism of import into mitochondria, pathways of assembly into the macromolecular enzyme complex. Recent advances in the elucidation of detailed structures of yeast F1 and F0 sectors have provided a better insight on structure and roles of each subunit of the multisubunit enzyme complex. In contrast to the ancentral bacterial enzymes which represents the simplest form of the enzyme, the yeast mitochondrial ATP synthase has included a number of additional subunits of the F0 sector during the course of evolution. It is now clear that these subunits are components of the stator stalk and/or are associated with the dimeric form of the enzyme complex. The dimerization of the mitochondrial ATP synthase is essential for proper development of mitochondrial innermembrane curvature known as cristae. This shows that mitochondrial ATP synthase is not only important for ATP production, but also crucial for the formation cristae, a characteristic morphology of mitochondria. Yeast therefore may be of used for understanding the molecular evolution eucaryotic mitochondrial ATP synthase. More comprehensive and detailed studies on the roles of the yeast supernumerary F0 subunits are needed. In addition, the subunit composition and catalytic mechanism of the yeast enzyme is very similar to that of human and other mammalian enzymes. Yeast may also provide a model for understanding human and other mammalian mitochondrial ATP synthases in both physiological and diseased states. The biogenesis of mitocondrial ATP synthase involves assembly of polypeptide subunits from dual genetic origin, the nuclear and mitochondrial genomes. Most of the subunits are expressed in the nuclear-cytosolic system and then imported into mitochondria. Future studies should also be directed to elucidation of how the expression of the two genomes is regulated and what factors affect the efficiency of subunit importation into mitochondria and their assembly into the functional enzyme complex. Improved understanding on subunits expression, import, and assembly my contribute to the development of genetic engineering strategies such as gene therapy to cure human mitochondrial genetic diseases. In yeast, several genes encoding mitochondrial ATP synthase subunits have successfully been expressed in the nuclear-cytosolic system followed by import and assembly into the mitochondrial ATP synthase complex. The activity of the enzyme is naturally regulated in order to prevent wasteful hydrolysis of ATP especially when the oxygen supply to the electron transport chain is limited. In yeast, INH protein has been shown to play roles as an F1-ATP synthase intrinsic inhibitor. Further studies are required to elucidate the regulation of the INH expression and its activity in relation to energy metabolism and growth conditions including oxygen deprivation, environmental changes etc. The detailed mechanism by which yeast INH protein regulates the activity of the ATP synthase needs to be revealed. Other cellular functions of the INH protein, such as in dimerization and oligomerization of the ATP synthase that affect the mitochondrial ultra structure and morphology, need to be described. Importantly, the potency of ATP synthase as a molecular drug target for development of antimicrobial and antitumor agents for human therapy, as well as development of pesticides, herbicides and insecticides for agriculture, may be explored using the yeast enzyme as a model.
References
Abraham JP, Leslie AGW, Lutter R, Walker JE (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370:621–628
Anselmi C, Davies KM, Faraldo-Gómez JD (2018) Mitochondrial ATP synthase dimers spontaneously associate due to a long-range membrane-induced force. J Gen Phys 150:763–770
Arnold I, Pfeiffers K, Neupert W, Stuart RA, Schagger H (1999) ATP synthase of yeast mitochondria. Isolation of subunit j and disruption of the ATP18 gene. J Biol Chem 174:36–40
Arselin G, Vaillier J, Graves PV, Velours J (1996) ATP synthase of yeast mitochondria. Isolation of the subunit h and disruption of the ATP14 gene. J Biol Chem 271:20284–20290
Artika IM (2007) Structural and functional analysis of FLAG tagged-subunit 8 of yeast Saccharomyces cerevisiae mitochondrial ATP synthase. Microbiol Indones 1:33–36
Artika IM (2009) Membrane topology of subunit 8 variant of yeast Saccharomyces cerevisiae mitochondrial ATP synthase. Microbiol Indones 3:37–41
Attardi G, Schatz G (1988) Biogenesis of mitochondria. Annu Rev Cell Biol 4:289–333
Backes S, Hess S, Boos F, Woellhaf MW, Gödel S, Jung M, Mühlhaus T, Herrmann JM (2018) Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J Cell Biol 217:1369–1382
Bateson MG, Devenish RJ, Nagley P, Prescott M (1999) Single copies of subunit b, oligomycin sensitivity conferring protein and d are present in the S. cerevisiae mitochondrial ATP synthase. J Biol Chem 274:7462–7466
Boyer PD (1975) A model for conformational coupling of membrane potential and proton translocation to ATP synthesis and active transport. FEBS Lett 58:1–6
Boyer PD (1989) A perspective of the binding change mechanism for ATP synthesis. FASEB J 3:2164–2178
Cheng MY, Harlt F, Martin J, Pollock RA, Kalousek F, Neupert W et al (1989) Mitochondrial heat-shock protein hsp60 is essential for assembly of protein imported into yeast mitochondria. Nature 337:620–625
Cox GB, Devenish RJ, Gibson F, Howitt SM, Nagley P (1992) The structure and assembly of ATP synthase. In: Ernster L (ed) Molecular mechanism in bioenergetics. Elsevier, Amsterdam, pp 283–315
Craig EA (2018) Hsp70 at the membrane: driving protein translocation. BMC Biol 16:1–11
Cross RL (1992) The reaction mechanism of F1F0-ATP synthases. In: Ernster L (ed) Molecular mechanism in bioenergetics. Elsevier, Amsterdam, pp 317–330
Cross RL, Duncan TM (1996) Subunit rotation in F1F0-ATP synthases as a mean of coupling proton transport through F0 to the binding changes in F1. J Bioenerg Biomembr 28:403–408
Dabbeni-Sala F, Rai AK, Lippe, G (2012) F0F1 ATP synthase: A fascinating challenge for proteomics, proteomics - human diseases and protein functions, Prof. Tsz Kwong Man (Ed.), ISBN: 978-953-307-832-8, InTech, Available from: http://www.intechopen.com/books/proteomics-humandiseases-and-protein-functions/f0f1-atp-synthase-a-fascinating-challenge-for-proteomics. Accessed 30 June 2018
Dautant A, Velours J, Giraud MF (2010) Crystal structure of the Mg.ADP-inhibited state of the yeast F1c10-ATP synthase. J Biol Chem 285:29502–29510
Davies KM, Anselmi C, Wittig I, Faraldo-Gómez JD, Kühlbrandt W (2012) Structure of the yeast F1F0-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci U S A 109:13602–13607
Devenish RJ, Prescott M, Roucou X, Nagley P (2000) Insights into ATP synthase assembly and function through the molecular genetic manipulation of subunits of the yeast mitochondrial enzyme complex. Biochim Biophys Acta 1458:428–442
Devenish RJ, Prescott M, Rodgers AJW (2008) The structure and function of mitochondrial F1F0-ATP synthase. Int Rev Cell Mol Biol 267:1–58
Dienhart M, Pfeiffer K, Schägger H, Stuart RA (2002) Formation of the yeast F1F0-ATP synthase dimeric complex does not require the ATPase inhibitor protein, Inh1. J Biol Chem 277:39289–39295
Faccenda D, Campanella M (2012) Molecular regulation of the mitochondrial F1F0-ATP synthase: physiological and pathological significance of the inhibitory factor 1 (IF1). Int J Cell Biol 2012:1–12
Fox TD (2012) Mitochondrial protein synthesis, import, and assembly. Genetics 192:1203–1234
Guo H, Bueler SA, Rubinstein JL (2017) Atomic model for the dimeric F0 region of mitochondrial ATPsynthase. Science 358:936–940
Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W, Meier T (2016) Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol Cell 63:445–456
Hartl F, Pfanner N, Nicholson DW, Neupert W (1989) Mitochondrial protein import. Biochem Biophys Acta 988:1–45
Hauckle V, Lithgow T (1997) The first steps of protein import into mitochondria. J Bioenerg Biomembr 29:11–17
He J, Ford HC, Carrol J, Douglas C, Gonzales E, Ding S, Fearnley IM, Walker JE (2018) Assembly of the membrane domain of ATP synthase in human mitochondria. Proc Natl Acad Sci U S A 115:2988–2993
Junge W, Lill H, Engelbrecht S (1997) ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biol Sci 22:420–423
Kabaleeswaran V, Puri N, Walker JE, Leslie AGW, Mueller DM (2006) Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J 25:5433–5442
Kresge N, Simoni RD, Hill RL (2006) ATP synthesis and the binding change mechanism: the work of Paul D. Boyer. J Biol Chem 281:e18–e20
Kühlbrandt W (2019) Structure and mechanism of F-type ATP synthases. Annu Rev Biochem 88:21.1–21.35
Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–148
Nagley P (1988) Eukaryote membrane genetics: the F0 sector of mitochondrial ATP synthase. Trends Genet 4:46–52
Noji H, Yasuda R, Yoshida M, Kinosita K (1997) direct observation of the rotation of F1-ATPase. Nature 386:299–302
Norais N, Prome D, Velours J (1991) ATP synthase of yeast mitochondria: characterization of subunit d and sequence analysis of the structural gene ATP7. J Biol Chem 266:16541–16549
Rak M, Gokova S, Tzagoloff A (2011) Modular assembly of yeast mitochondrial ATP synthase. EMBO J 30:920–930
Roucou X, Artika IM, Devenish RJ, Nagley P (1999) Bioenergetic and structural consequences of allotopic expression of subunit 8 of yeast mitochondrial ATP synthase: the hydrophobic character of residues 23 and 24 is essential for maximal activity and structural stability of the enzyme complex. Eur J Biochem 261:444–451
Rubinstein JL, Walker JE (2002) ATP synthase from Saccharomyces cerevisiae: location of the OSCP subunit in the peripheral stalk region. J Mol Biol 321:613–619
Rubinstein JL, Dickson VK, Runswick MJ, Walker JE (2005) ATP synthase from Saccharomyces cerevisiae: location of subunit h in the peripheral stalk region. J Mol Biol 345:513–520
Sebald W, Hoppe J (1981) Proteolipid subunit of ATP synthase complex. Curr Top Bioeneg 12:1–64
Senior AE, Weber J, Al-Shawi MK (1995) Catalytic mechanism of Escherichia coli F1-ATPase. Biochem Soc Trans 23:747–752
Sokol AM, Sztolsztener ME, Wasilewski M, Heinz E, Chacinska A (2014) Mitochondrial protein translocases for survival and wellbeing. FEBS Lett 588:2484–2495
Song J, Pfanner N, Becker T (2018) Assembling the mitochondrial ATPsynthase. Proc Natl Acad Sci U S A 115:2850–2852
Spannagel C, Vaillier J, Arselin G, Graves P, Velours J (1997) The subunit f of mitochondrial yeast ATP synthase. Characterization of the protein and disruption of the structural gene ATP17. Eur J Biochem 247:1111–1117
Srivastava AP, Luo M, Zhou W, Symersky J, Bai D, Chambers MG, Faraldo-Gómez JD, Liao M, Mueller DM (2018) High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 360:1–8
Stephens AN, Roucou X, Artika IM, Devenish RJ, Nagley P (2000) Topology and proximity relationships of yeast mitochondrial ATP synthase subunit 8 determined by unique introduced cysteine residues. Eur J Biochem 267:6443–6451
Stephens AN, Nagley P, Devenish RJ (2003) Each yeast mitochondrial F1F0-ATP synthase complex contains a single copy of subunit 8. Biochim Biophys Acta 1607:181–189
Stewart AG, Laming EM, Sobti M, Stock D (2014) Rotary ATPases: dynamic molecular machines. Curr Opin Struct Biol 25:40–48
Su CH, McStay GP, Tzagolof A (2014) Assembly of the rotor component of yeast mitochondrial ATP synthase is enhanced when Atp9p is supplied by Atp9p-Cox6p complexes. J Biol Chem 289:31605–31616
Symersky J, Osowski D, Walters DE, Mueller DM (2012a) Oligomycin frames a common drug-binding site in the ATP synthase. Proc Natl Acad Sci U S A 109:13961–13965
Symersky J, Pagadala V, Osowski D, Krah A, Meier T, Faraldo-Gómez JD, Mueller DM (2012b) Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol 19:485–505
Ting SY, Schilke BA, Hayashi M, Craig EA (2014) Architecture of the TIM23 inner mitochondrial translocon and interactions with the matrix import motor. J Biol Chem 289:28689–28696
Tzagoloff A, Myers AM (1986) Genetics of mitochondrial biogenesis. Annu Rev Biochem 55:249–285
Vaillier J, Arselin G, Graves P, Carmougrand N, Velours J (1999) Isolation of supernumerary yeast ATP synthase subunit e and i. Characterization of subunit i and disruption of its structural gene ATP18. J Biol Chem 274:545–548
Wagner K, Perschil I, Fichter CD, van der Laan M (2010) Stepwise assembly of dimeric F1F0-ATP synthase in mitochondria involves the small Fo-subunits k and i. Mol Biol Cell 21:1494–1504
Wiedemann N, Pfanner N (2017) Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86:685–714
Xu T, Pagadala V, Mueller DM (2015) Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microbial Cell 2:105–125
Zhou A, Rohou A, Schep DG, Bason JV, Montgomery MG, Walker JE et al (2015) Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. eLife 4:e10180. https://doi.org/10.7554/eLife.10180
Acknowledgements
This work was carried out without external funding support. The author thanks Dr. John Acton for his assistance at the manuscript stage.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Artika, I.M. Current understanding of structure, function and biogenesis of yeast mitochondrial ATP synthase. J Bioenerg Biomembr 51, 315–328 (2019). https://doi.org/10.1007/s10863-019-09809-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-019-09809-4