
Abstract
Heat shock protein 90 (Hsp90) is an attractive therapeutic target. Geldanamycin (GA), the first identified Hsp90 inhibitor, exhibited potent antitumor activity, but possessed significant hepatotoxicity. To overcome the hepatotoxicity derived from the quinone structure of GA, a non-quinone GA derivative 17-demethoxy-reblastatin (17-DR) was obtained from a genetically modified strain of Streptomyces hygroscopicus. In the present study, we examined the anticancer effects of 17-DR on human hepatocellular carcinoma (HCC) cell lines HepG2 and SMMC7721, and its underlying mechanisms. The results indicated that 17-DR could concentration-dependently inhibit the proliferation, and decrease the colony formation in HCC cells. It also induced significant apoptosis in HCC cells, which was mediated by mitochondria via a caspase-dependent pathway. The mechanisms involved in 17-DR-induced apoptosis included the downregulation of myeloid cell leukemia-1 (Mcl-1), and upregulation of Bcl-2 antagonist killer 1 (Bak). And the upregulated Bak expression resulted from downregulation of Mcl-1 played an essential role in this process. Taken together, these results indicated that 17-DR possessed potent anticancer effects on HCC cells by inhibiting cell proliferation and inducing apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third most frequent cause of cancer-related mortality globally, with 782,000 new cases occurring and 746,000 deaths from liver cancer in 2012 worldwide (Flores and Marrero 2014). The incidence and mortality rates continue to rise all over the world. To date, surgery is still the major treatment for HCC. However, many patients are not suitable to receive surgery due to various reasons, such as distant metastasis, unresectable tumors, insufficient hepatic function and poor health conditions. Moreover, HCC has a high recurrence rate after resection. Therefore, neoadjuvant therapy is usually applied to treat HCC patients (Zhuo et al. 2013). It is well known that chemotherapy is a vital management for advanced HCC (Cervello et al. 2012). Conventional cytotoxic therapies may result in obvious morbidity in patients with solid tumors since they simultaneously affect rapidly dividing malignant and normal cells, therefore undermining the survival of normal cells (Zhang et al. 2014). As for the targeted drugs, sorafenib, a multikinase inhibitor, is currently the only targeted therapeutic agent approved for the treatment of advanced HCC (Gauthier and Ho 2013). However, patients usually develop resistance after sorafenib treatment. Therefore, identification of new drugs targeting different signaling pathways is urgently needed for the treatment of HCC (Zheng et al. 2014).
Heat shock protein 90 (Hsp90) is a molecular chaperone playing key roles in the preservation of the conformation, stability and function of various signaling proteins involved in the pathways of cell proliferation, cell cycle progression, invasion, metastasis and angiogenesis. Notably, most of its client proteins are oncogenic proteins, such as human epidermal growth factor receptor-2 (Her2), epidermal growth factor receptor (EGFR), protein kinase B (Akt) and matrix metalloproteinase (MMP), which make it an attractive target for the development of antitumor agents (Kamal et al. 2004; Zhang and Burrows 2004; Taipale et al. 2010; Da Silva and Ramos 2012). In particular, Hsp90 is constitutively expressed at 2‑10‑fold higher levels in cancer cells, and its inhibitors possess remarkable selectivity for tumor cells, as compared with normal cells (Neckers et al. 1999; Kamal et al. 2003). It has been fully documented that Hsp90 inhibitors may simultaneously affect several abnormal signaling pathways in cancer cells and can also overcome the drug resistance of cancer cells (Chen et al. 2014). Geldanamycin (GA), a benzoquinone ansamycin antibiotic (Fig. 1a), identified as the first Hsp90 inhibitor, binds to the adenosine tri-phosphate (ATP) pocket in the N-terminal domain of Hsp90 and inhibits its ATP-dependent chaperone functions (Whitesell et al. 1994; Kim et al. 2013). The antitumor potential of GA has been studied for a long time, clinical evaluation of GA has not been performed due to its hepatotoxicity and poor solubility (Supko et al. 1995). To overcome the hepatotoxicity derived from the quinone structure of GA, a non-quinone GA derivative, 17-demethoxy-reblastatin (17-DR) was obtained from a genetically modified strain of Streptomyces hygroscopicus JCM4427 (Shin et al. 2008). 17-DR displayed stronger inhibitory activity to yeast Hsp90 ATPase (IC50, 1.82 μM) compared with the original Hsp90 inhibitor GA (IC50, 3.19 μM), and anticancer effects in human breast cancer cells (Wu et al. 2012; Zhao et al. 2014).

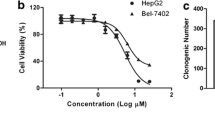
Inhibitory effects of 17-DR on the viability of HepG2 and SMMC7721 cells. a Chemical structure of GA and 17-DR. b HepG2 and SMMC7721 cells were treated with different concentrations of 17-DR (12.5, 25, 50, 100, 200 μM) or DMSO for 48 h. Cell viability was measured by MTT assay. *p < 0.05 and **p < 0.01 versus no 17-DR treatment. c Cells were treated with, i DMSO, ii 2.5 μM 17-DR, iii 5 μM 17-DR, iv) 10 μM 17-DR for 7 days, then the colonies were stained with crystal violet
In the current study, we explored the effects of 17-DR on the cell proliferation and apoptosis in human HCC cell lines HepG2 and SMMC7721, and its underlying mechanisms.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), RPMI-1640 medium and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). GA, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) and propidium iodide (PI) were from Sigma (St. Louis, Missouri, USA). The cell-permeable general caspase inhibitor z-VAD-fmk was purchased from Calbiochem (La Jolla, CA, USA). Primary antibodies for B-cell lymphoma-2 (Bcl-2), Bcl-2 antagonist/killer 1 (Bak), Bcl-2-associated X protein (Bax), and myeloid cell leukemia-1 (Mcl-1) were obtained from ProteinTech (Chicago, IL, USA), poly (ADP-ribose) polymerase (PARP) and β-actin from Santa Cruz (Santa Cruz, CA, USA). 17-DR was obtained as described previously, its structure was shown in Fig. 1a (Zhao et al. 2014).
Cell lines and cell culture
The human normal liver cells L-02, human HCC cell lines HepG2 and SMMC7721 were obtained from Shanghai Cell Bank (Shanghai, China) and cultured in our laboratory. HepG2 and SMMC7721 cells were cultured in DMEM and L-02 cells in RPMI-1640 medium supplemented with 10 % FBS, 1 × 105 U/L penicillin, and 100 mg/L streptomycin. These cells were maintained at 37 °C in a 5 % CO2 humidified atmosphere.
Cell viability assay
Cell viability was determined by MTT assay. The HCC cells were plated in triplicate at 6 × 103 cells/well in 96-well culture plates and treated with different concentrations of 17-DR (0, 12.5, 25, 50, 100, 200 μM) or GA (0, 2.5, 5, 10, 20, 40 μM). L-02 cells were plated in triplicate at 9 × 103 cells/well in 96-well culture plates and treated with 17-DR at the concentration of 0, 25, 50, 100, 200, 400 μM or GA at the concentration of 0, 5, 10, 20, 40, 80 μM. 48 h later, 15 μL MTT (5 mg/mL) was added to each well for an additional 4 h. The blue MTT formazan precipitate was then dissolved in 150 μL DMSO. The absorbance at 570 nm was measured on a scanning multi-well spectrophotometer. Cell viability was expressed as a percentage of control.
Colony formation assay
The cells were seeded in 6-well culture plates at 1 × 104 cells/well and allowed to attach overnight, then treated with DMSO, 17-DR (2.5, 5, 10 μM) under standard cell culture conditions for 7 days. After washing twice with phosphate buffer saline (PBS), the colonies were fixed with ice-cold methanol for 30 min, stained with 0.5 % crystal violet for 10 min, washed with distilled water, dried at room temperature, and visualized by a camera.
Analysis of apoptosis by flow cytometry with PI staining
HCC cells were seeded at 4 × 105 cells/well in 6-well culture plates and allowed to reach exponential growth for 24 h before treatment. The cells were treated with different concentrations of 17-DR (0, 25, 50, and 100 μM) for 48 h, subjected to PI staining, and then evaluated the sub-G1 deoxyribonucleic acid (DNA) content on a BD Accuri C6 flow cytometer.
Mitochondrial membrane potential (ΔΨm) assay using fluorescent microscope
The cells were seeded at 2 × 105 cells/well in 12-well culture plates and allowed to attach overnight before treatment. Changes in ΔΨm after different treatments were evaluated by staining with the cationic dye tetrechloro-tetraethylbenzimidazol carbocyanine iodide (JC-1) according to the manufacturer’s instruction (Beyotime Institute of Biotechnology, China). After the incubation, the dye was aspirated from the plates, and the plates were washed three times with 1 × JC-1 buffer and examined with an invert fluorescent microscope (Olympus IX71, Japan) using both red and green channels.
Western blot analysis
HCC cells were plated at 5 × 105 cells/well in 6-well culture plates. After incubation with 17-DR for 24 h, the cells were harvested, washed twice with ice-cold PBS, and lysed in radio immunoprecipitation assay (RIPA) buffer for 30 min on ice. The whole-cell lysates were separated on a 12 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking in 5 % skim milk, the membrane was incubated overnight with appropriate primary antibodies at 4 °C, then incubated with the corresponding secondary antibodies. Labeled bands were detected by an enhanced chemiluminescence substrate (Millipore, USA), and imaged with a gel imaging equipment (Bio-Rad, USA).
Plasmid transfection
The pCMV-HA-Mcl-1 plasmid and control plasmid were obtained from GenePharma (China). 1 μg plasmid was transiently transfected into HCC cells in 6-well culture plates using 5 μL Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen, USA). After 24 h of incubation, whole-cell lysates were prepared for immunoblot analysis.
Statistical analysis
Data are presented as mean ± SD. All experiments in this paper were repeated at least three times. Statistical analysis was performed using one-way analysis of variance followed by LSD test with SPSS 16.0 software. A p-value less than 0.05 was considered statistically significant. Significant differences are indicated by *p < 0.05, and **p < 0.01.
Results
Effects of 17-DR on cell proliferation
The antiproliferative effects of 17-DR on HCC cells were evaluated by MTT assay. The results demonstrated that 17-DR displayed notable antiproliferative activities on HCC cells in a concentration-dependent manner, with IC50 values of 51.3 and 57.2 μM for HepG2 and SMMC7721 cells, respectively, after treatment for 48 h (Fig. 1b). Then we observed the effects of 17-DR at lower concentrations on the colony formation of HCC cells. 17-DR also showed obvious cytotoxicities against the HCC cells (Fig. 1c).
In order to evaluate the efficacy and toxicity of 17-DR preliminarily, GA was used as the control. As a result, GA exhibited obvious cytotoxicity to HCC cells, with IC50 values of 7.6 and 8.4 μM for HepG2 and SMMC7721 cells, respectively, after treatment for 48 h (Fig. S1). As regards the cytotoxicity to normal liver cells L-02, both GA and 17-DR displayed less killing activity. The IC50 value of GA was 23.8 μM after treatment L-02 cells for 48 h (Fig. S2a), while the IC50 value of 17-DR could not be calculated within the concentrations of 0–400 μM after treatment for 48 h (Fig. S2b).
17-DR induces apoptosis in HCC cells
It has been well established that most of the currently used chemotherapeutic drugs exert their antitumor effects by inducing apoptosis in neoplastic cells (Dean et al. 2012). We examined 17-DR-induced apoptosis by flow cytometry with PI staining. Compared with DMSO-treated cells, HepG2 and SMMC7721 cells treated with 17-DR at the concentration of 25, 50, 100 μM presented higher apoptotic rates (Fig. 2a and b). In association with this, ΔΨm was detected by JC-1 staining. The 17-DR-treated cells exhibited a marked reduction in red fluorescence while increased signals in green fluorescence, indicating reduction in ΔΨm occurred and 17-DR-induced apoptosis in HCC cells was mediated by mitochondria (Fig. 2c). To verify whether caspases play a role in apoptosis induced by 17-DR, we applied the cell-permeable general caspase inhibitor z-VAD-fmk to treat the HCC cells. As a result, z-VAD-fmk treatment inhibited 17-DR-induced cytotoxicity, suggesting a caspase-dependent killing mechanism (Fig. 2d). Taken together, 17-DR induced apoptosis which was mediated by mitochondria via a caspase-dependent pathway in HCC cells.

17-DR induces apoptosis in HCC cells. a and b HepG2 and SMMC7721 cells were treated with 17-DR (25, 50, 100 μM) or DMSO for 48 h. Apoptosis was examined by flow cytometry with PI staining. **p < 0.01 versus no 17-DR treatment. c Cells were treated with 17-DR (25, 50, 100 μM) or DMSO for 24 h. ΔΨm was measured by JC-1 staining. d Cells were treated with the general caspase inhibitor z-VAD-fmk (20 μM) for 1 h before the addition of 17-DR (50 μM) for a further 48 h. Cell viability was assessed by MTT assay. **p < 0.01 versus no z-VAD-fmk treatment
Bcl-2 family proteins are involved in 17-DR-induced apoptosis
Apoptosis mediated by the mitochondrial pathway is regulated by the Bcl-2 family of proteins (Leanza et al. 2012). To elucidate the mechanisms involved in 17-DR-induced apoptosis in HCC cells, the expression levels of antiapoptotic proteins Bcl-2, Mcl-1, and proapoptotic proteins Bak, Bax were analyzed by immunoblotting in the HCC HepG2 and SMMC7721 cells. As shown in Fig. 3a–c, the antiapoptotic protein Mcl-1 was downregulated, the proapoptotic protein Bak was upregulated, while Bcl-2 was modestly downregulated and Bax remained unchanged. In addition, cleavage of the caspase-3 substrate PARP was observed after exposure to 17-DR treatment, which also confirmed that apoptotic process occurred in HCC cells (Fig. 3a–c).

Involvement of Bcl-2 family proteins and PARP in x17-DR-induced apoptosis. a-c HepG2 and SMMC7721 cells were treated with 17-DR (25, 50, 100 μM) or DMSO for 24 h. Whole-cell lysates were subjected to examine Bak, Bax, Bcl-2, Mcl-1, and PARP protein levels by Western blot analysis. β-actin was used as a loading control
Overexpression of Mcl-1 protects HCC cells from apoptosis
The antiapoptotic protein Mcl-1 is considered to preferentially inhibit the activation of Bak, whereas Bcl-2 inactivate Bax (Pearce and Lyles 2009). Because Bak was significantly upregulated after 17-DR treatment, then we examined the role of Mcl-1 in 17-DR-induced apoptosis by transfecting complementary DNA (cDNA) encoding Mcl-1 into HepG2 and SMMC7721 cells (Fig. 4a). The overexpression of Mcl-1 significantly protected the cells from apoptosis induced by 17-DR (Fig. 4b and c). The expression of Bak was downregulated in the Mcl-1-overexpressing HCC cells after 17-DR treatment, compared with cells transfected with vector alone (Fig. 4d). Altogether, these results confirmed that Mcl-1 played an important role in 17-DR-induced apoptosis.

Overexpression of Mcl-1 protects HCC cells from apoptosis. a HepG2 and SMMC7721 cells were transiently transfected with vector alone or Mcl-1 cDNA. 24 h later, the whole-cell lysates were subjected to Western blot analysis. b and c HepG2 and SMMC7721 cells were transiently transfected with vector alone or Mcl-1 cDNA. 24 h later, cells were treated with 17-DR (50 μM) or DMSO for a further 48 h before apoptosis was measured by flow cytometry with PI staining. **p < 0.01 versus no Mcl-1 cDNA treatment. d HepG2 and SMMC7721 cells were transiently transfected with vector alone or Mcl-1 cDNA. 24 h later, cells were treated with 17-DR (50 μM) or DMSO for a further 24 h. Whole-cell lysates were subjected to Western blot analysis
Discussion
Apoptosis is an important cellular process for maintaining the homeostasis between cell proliferation and death, and is also vital for the removal of damaged, infamed, and diseased cells (Liu and Wang 2012). Deregulated apoptosis is often considered a hallmark of cancer (Hanahan and Weinberg 2000). Apoptosis can be initiated through two distinct yet correlated pathways, the mitochondrial/intrinsic and extrinsic pathway (Quinn et al. 2011). Most anticancer agents induce apoptosis through the mitochondrial pathway, which is triggered in response to stress signals, such as DNA damage or endoplasmic reticulum (ER) stress, caused by chemotherapy, oncogene activation, growth factor deprivation, infection, hypoxia or ultraviolet radiation (Mohana-Kumaran et al. 2014). The Bcl-2 family, which includes both antiapoptotic and proapoptotic members, tightly regulates the mitochondrial apoptotic pathway (Mohana-Kumaran et al. 2014). The antiapoptotic proteins such as Mcl-1and Bcl-2 protect mitochondrial integrity, whereas the proapoptotic members of the family such as Bak and Bax promote the release of apoptogenic proteins, such as cytochrome c, apoptosis inducing factor (AIF), and second mitochondria-derived activator of caspase (Smac/DIABLO) from the mitochondria to the cytosol (Martinou and Green 2001; Adams and Cory 1998). The released apoptogenic proteins induce the activation of specific proteinases termed caspases, which cleave several vital proteins provoking the cell death (Green 2005). The ratio of proapoptotic to antiapoptotic protein is crucial to regulate the apoptotic process.
In this work, the mechanisms involved in 17-DR-induced apoptosis were evaluated by antiapoptotic and proapoptotic protein expression levels in the HCC HepG2 and SMMC7721 cells. We found that 17-DR treatment decreased the level of antiapoptotic protein Mcl-1, and increased the level of proapoptotic Bak, so the ratio of Mcl-1/Bak was decreased. At the molecular level, proapoptotic proteins of the Bcl-2 family, Bak and Bax, play primary roles in the mitochondrial apoptotic pathway induced by various apoptotic stimuli downstream from BH3-only proteins (Shimazu et al. 2007). The antiapoptotic protein Mcl-1 is considered to preferentially inhibit the activation of Bak, whereas Bcl-2 inactivate Bax (Pearce and Lyles 2009). Overexpression of Mcl-1 protected HCC cells from apoptosis induced by 17-DR, and resulted in decreased Bak expression. Thus 17-DR-induced apoptosis was mediated by upregulated Bak expression resulted from downregulation of Mcl-1 through the mitochondrial apoptotic pathway.
In conclusion, 17-DR induced significant apoptosis which was primarily triggered by downregulation of Mcl-1 expression through the mitochondrial apoptotic pathway in HCC cells.
References
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Cervello M, Mccubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G (2012) Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget 3:236–260
Chen D, Shen A, Li J, Shi F, Chen W, Ren J et al (2014) Discovery of potent N-(isoxazol-5-yl)amides as HSP90 inhibitors. Eur J Med Chem 87:765–781
Da Silva VC, Ramos CH (2012) The network interaction of the human cytosolic 90 kDa heat shock protein Hsp90: a target for cancer therapeutics. J Proteomics 75:2790–2802
Dean E, Greystoke A, Ranson M, Dive C (2012) Biomarkers of cell death applicable to early clinical trials. Exp Cell Res 318:1252–1259
Flores A, Marrero JA (2014) Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clin Med Insights Oncol 8:71–76
Gauthier A, Ho M (2013) Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Res 43:147–154
Green DR (2005) Apoptotic pathways: ten minutes to dead. Cell 121:671–674
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425:407–410
Kamal A, Boehm MF, Burrows FJ (2004) Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med 10:283–290
Kim T, Keum G, Pae AN (2013) Discovery and development of heat shock protein 90 inhibitors as anticancer agents: a review of patented potent geldanamycin derivatives. Expert Opin Ther Pat 23:919–943
Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabo I (2012) Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med 4:577–593
Liu Q, Wang HG (2012) Anti-cancer drug discovery and development: Bcl-2 family small molecule inhibitors. Commun Integr Biol 5:557–565
Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2:63–67
Mohana-Kumaran N, Hill DS, Allen JD, Haass NK (2014) Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment Cell Melanoma Res 27:525–539
Neckers L, Mimnaugh E, Schulte TW (1999) Hsp90 as an anti-cancer target. Drug Resist Updat 2:165–172
Pearce AF, Lyles DS (2009) Vesicular stomatitis virus induces apoptosis primarily through Bak rather than Bax by inactivating Mcl-1 and Bcl-XL. J Virol 83:9102–9112
Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S et al (2011) Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs 20:1397–1411
Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y et al (2007) NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev 21:929–941
Shin JC, Na Z, Lee DH, Kim WC, Lee K, Shen YM et al (2008) Characterization of tailoring genes involved in the modification of geldanamycin polyketide in Streptomyces hygroscopicus JCM4427. J Microbiol Biotechnol 18:1101–1108
Supko JG, Hickman RL, Grever MR, Malspeis L (1995) Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 36:305–315
Taipale M, Jarosz DF, Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11:515–528
Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (1994) Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 91:8324–8328
Wu CZ, Jang JH, Ahn JS, Hong YS (2012) New geldanamycin analogs from Streptomyces hygroscopicus. J Microbiol Biotechnol 22:1478–1481
Zhang H, Burrows F (2004) Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 82:488–499
Zhang W, Liu S, Liu K, Ji B, Wang Y, Liu Y (2014) Knockout of ADAM10 enhances sorafenib antitumor activity of hepatocellular carcinoma in vitro and in vivo. Oncol Rep 32:1913–1922
Zhao Q, Wu CZ, Lee JK, Zhao SR, Li HM, Huo Q et al (2014) Anticancer effects of the Hsp90 inhibitor 17-demethoxy-reblastatin in human breast cancer MDA-MB-231 cells. J Microbiol Biotechnol 24:914–920
Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP (2014) KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 74:487–495
Zhuo L, Liu J, Wang B, Gao M, Huang A (2013) Differential miRNA expression profiles in hepatocellular carcinoma cells and drug-resistant sublines. Oncol Rep 29:555–562
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81372899, 81302671), the Natural Science Foundation of Anhui Province (1408085QH162), and the Natural Science Foundation of Bengbu Medical College (BYKY1408ZD).
Conflicts of interest
The authors declare that they have no conflict of interests regarding the publication of this paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Surong Zhao and Hongmei Li contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Proliferation of HCC cells inhibited by GA. HepG2 and SMMC7721 cells were treated with different concentrations of GA (2.5, 5, 10, 20, 40 μM) or DMSO for 48 h. Cell viability was measured by MTT assay. **p < 0.01 versus no GA treatment (TIFF 730 kb)
Fig. S2
Effects of GA and 17-DR on the viability of normal liver cells. a L-02 cells were treated with different concentrations of GA (5, 10, 20, 40, 80 μM) or DMSO for 48 h. b L-02 cells were treated with different concentrations of 17-DR (25, 50, 100, 200, 400 μM) or DMSO for 48 h. Cell viability was measured by MTT assay. *p < 0.05 and **p < 0.01 versus no GA or 17-DR treatment. (TIFF 991 kb)
Rights and permissions
About this article

Cite this article
Zhao, S., Li, H., Jiang, C. et al. 17-Demethoxy-reblastatin, an Hsp90 inhibitor, induces mitochondria-mediated apoptosis through downregulation of Mcl-1 in human hepatocellular carcinoma cells. J Bioenerg Biomembr 47, 373–381 (2015). https://doi.org/10.1007/s10863-015-9620-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-015-9620-1