
Abstract
The in vitro and in vivo performance of hydroxyapatite (HAp) coatings can be modified by the addition of different trace ions, such as silicon (Si), lithium (Li), magnesium (Mg), zinc (Zn) or strontium (Sr) into the HAp lattice, to more closely mirror the complex chemistry of human bone. To date, most of the work in the literature has considered single ion-substituted materials and coatings, with limited reports on co-substituted calcium phosphate systems. The aim of this study was to investigate the potential of radio frequency magnetron sputtering to deposit Sr and Zn co-substituted HAp coatings using Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The FTIR and XPS results highlight that all of the Sr, Zn and Sr-Zn co-substituted surfaces produced are all dehydroxylated and are calcium deficient. All of the coatings contained HPO4 2− groups, however; only the pure HAp coating and the Sr substituted HAp coating contained additional CO3 2− groups. The XRD results highlight that none of the coatings produced in this study contain any other impurity CaP phases, showing peaks corresponding to that of ICDD file #01-072-1243 for HAp, albeit shifted to lower 2θ values due to the incorporation of Sr into the HAp lattice for Ca (in the Sr and Sr-Zn co-substituted surfaces only). Therefore, the results here clearly show that RF magnetron sputtering offers a simple means to deliver Sr and Zn co-substituted HAp coatings with enhanced surface properties.
Graphical Abstract
(a) XRD patterns for RF magnetron sputter deposited hydroxyapatite coatings and (b)-(d) for Sr, Zn and Sr-Zn co-substituted coatings, respectively. The XPS spectra in (b) confirms the presence of a HA sputter deposited coating as opposed to (c) XPS spectra for a Sr-Zn co-substituted sputter deposited coating.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Hydroxyapatite (HAp, Ca10(PO4)6(OH)2) is a highly valuable bone repair and regeneration material because of its similarity to the inorganic phase of human bone. It is bioactive, osteoconductive and has the ability to form a direct chemical bond with human bone [1–3]. In addition to the bulk form, HAp has been widely used as a coating material for orthopaedic implants in a variety of applications, and typically the release of Ca2+ and PO4 3− ions into the surrounding environment leads to the development of excellent interfacial strength between the implant and bone [3–6]. However, chemically pure synthetic HAp has a Ca/P ratio of 1.67, whereas biological apatite has variable stoichiometry and can contain various different calcium phosphate (CaP) phases and ionic substitutions, resulting in a non-stochiometric multi-element substituted material [7]. As such, when designing CaP biomaterials for clinical use one proposed route would be to introduce ionic substitutions into HAp in order to mimic the complex chemistry of human bone and thereby improve the biological performance of such materials, both in vitro and in vivo [8, 9].
A wide range of different ions such as Sr, Zn, Ag, Mg, CO3 2−, F and Si (amongst others) have been strategically incorporated into the structure of synthetic HAp, replacing Ca (or PO4 3−), as highlighted in Table 1 [10–48]. The substitution of such ions into pure HAp alters the crystal structure (crystallinity and crystal size) and also changes the properties of the material; namely the thermal and phase stability, its solubility, surface reactivity and its ability to be absorbed [49–51]. Particular attention is focused on several biologically active ions as Zn and Sr, which are naturally present in native bone tissue and can lead to advantageous effects on the biological response of osteoblasts. Sr and Zn both promote the osteoblast response and stimulate formation of new bone more readily than pure HAp [8, 9, 51, 52]. Sr is a beneficial trace element in human body and is shown to play an important role in bone formation. Sr stimulates a range of biological effects on the bone, such as anti-resorptive activity, osteoclast apoptosis and osteoblast stimulation [6, 8, 9, 49, 53–55]. These effects increase bone formation and reduce bone resorption, as well as improving bone strength, bone healing and microarchitecture and could potentially help in the treatment of osteoporosis [54, 56–58]. Zn is known to be an essential trace element in the body [9]. Zn is known to stimulate bone formation [8, 9, 59], but it also inhibits bacterial growth at the surgical site and improves the wound healing process [8, 52]. In addition, Zn also is known for its anti-inflammatory properties within the human body [58, 59], and its deficiency has been reported to reduce bone density and ductility, resulting in an increased probability of fracture [8, 9].
To date, there is a significant body of research dealing with single ion substituted CaP materials (as highlighted in Table 1). However, there are increasing numbers of reports on the use of co-substituted materials systems in both bulk powder form and coatings [10, 13, 14, 24, 26, 32, 58–62]. It has been suggested in the literature that co-substituted CaP systems may add significant value when considered for use in vivo (when compared to pure HAp and single ion substituted systems). A number of different deposition methods have already been investigated to produce co-substituted Ca-P coatings, including electrochemical deposition [19, 34], plasma spraying [63], sol-gel methods [32] and co-blasting [64]. Of the alternative coating technologies available, radio frequency (RF) magnetron sputtering has been shown to be particularly useful for the deposition of CaP coatings due to the ability of the technique to provide greater control of the coating’s properties and improved adhesion between the substrate and the coating [65–70]. With respect to producing co-substituted CaP coatings, RF magnetron sputtering allows co-deposition of multiple different target materials simultaneously and therefore provides an alternative and simple method to produce co-substituted CaP coatings [71–73]. Several reports in the literature highlight how it has already been used to deposit Sr or Si substituted HAp coatings [70–73]. However, to date, to the best of the authors’ knowledge, there is no available information on the utilisation of RF magnetron sputtering as a method to deposit Sr and Zn co-substituted CaP surfaces, with the primary objective of creating a surface whereby the concentrations of the different ion substitutions can be carefully controlled in line with requirements.
The present work was undertaken in order to study the deposition of Sr and Zn co-substituted Ca-P coatings from a custom designed RF magnetron sputtering facility utilising two sputtering targets (referred to as sources). Sr and Zn were chosen for this study as their combined application in a CaP coating could add significant value to such surfaces when compared to applying the ion substitutions separately. In particular, the influence of different target configurations on the properties of the Sr and Zn co-substituted sputter deposited coatings produced at a low discharge power level (150 W) were investigated. A low discharge power level was chosen for this study as the quality and consistency of the targets used could be guaranteed throughout the sputter deposition runs. All of the coatings produced were characterised after post-deposition annealing to 500 °C using Fourier Transform Infrared Spectroscopy (FTIR), X-Ray Diffraction (XRD) and X-Ray Photoelectron Spectroscopy (XPS).
2 Materials and methods
2.1 Synthesis and characterization of pure and ion-substituted HAp powders
HAp and HAp substituted with 10wt% Sr ([Sr/Ca + Sr)] and 10wt% Zn ([Zn/Ca + Zn)] powders were synthesized through a standard neutralization route. The experimental ratios of Ca/P or (Ca + M)/P (where M = Sr or Zn) were maintained at 1.67. Starting suspensions were prepared by dissolving Ca(NO3)2·4H2O (puriss. p.a., Sigma-Aldrich, Germany) and Sr(NO3)2 (puriss. p.a., Sigma-Aldrich, Germany) or Zn(NO3)2·6H2O (98%, reagent-grade, Sigma-Aldrich, Germany) in deionised H2O. An aqueous solution of H3PO4 (75%, puriss, Sigma-Aldrich, Germany) was utilised as a source of (PO4 3−) and was added to the starting suspensions at a slow addition rate (~0.75 ml/min). The pH of the synthesis mixture was kept around 9, using 25% ammonia solution. The synthesis temperature was kept constant at 45 °C. The precipitates obtained were left overnight to mature, filtered, and then washed with deionised H2O. Finally, the precipitates were dried at 105 °C for ~24 h, milled and selectively sieved to obtain powders with particle size less than 100 µm. The HAp powders were labelled HAp, 10SrHAp and 10ZnHAp, respectively.
The chemical constitutions of the prepared powders were determined by atomic absorption spectrometry (AAS) and energy dispersive X-ray spectrometry (EDX) utilising the following equipment: Varian SpektrAA 880 and Mira/LMU/Inca Energy 350, respectively. The molar ratios of (Ca + Sr)/P and (Ca + Zn)/P were calculated from the results obtained. Different functional groups were determined using Fourier transform infrared spectroscopy (FT-IR, Varian 800, Scimitar Series). The spectra were recorded in the range of 400–4000 cm−1 with spectral resolution 4 cm−1 with 30 scans co-added. The phase composition and crystallinity was analyzed by X-ray diffraction (XRD) utilising a Rigaku Ultima+ XRD system operating at 40 KV and 30 mA using Cu Kα (λ = 1.5406 Å) radiation. Scans was recorded over the range from 2Ө = 5° to 60°. For the phase identification the International Centre for Diffraction Data (ICDD) was used (card #01-072-1243 for HAp). The Brunauer–Emmett–Teller (BET) method was used to determine the specific surface area (SSA) of precipitated powders by N2 absorption (Quadrasorb-SI Kr, Quantachrome). The samples were degassed at 40 °C for 24 h prior the analyses. The values of mean sizes of particles of as-synthesized powders were estimated from the N2 adsorption isotherms using the BET particle diameter (d BET) from the following Eq. (1) by assuming the primary particles to be spherical:
where q is theoretical density of HAp—3.16 g/cm3 (according to ISO 13175-3:2012 Implants for surgery—Calcium phosphates—Part 3: Hydroxyapatite and beta-tricalcium phosphate bone substitutes).
2.2 Production and characterization of pure and ion-substituted HAp coatings
For this study coupons of chemically pure titanium (cpTi), Titanium International Ltd. (15 mm × 15 mm × 0.5 mm) were abraded using a succession of 800, and 1200 grade SiC papers. The coupons were twice sonicated for 10 min consecutively in acetone, isopropyl alcohol and distilled de-ionised water. The abraded coupons were then dried thoroughly in a convection oven at 70 °C for 12 h. Radio Frequency (RF) magnetron sputtering of the HAp and substituted HAp coatings was performed using a cluster of two high vacuum Torus 3 M sputtering sources in a custom designed system (Kurt J. Lesker Ltd, USA) each operating with a 13.56 MHz RF generator and an impedance matching network (Huettinger, GmbH, Germany). The coating nomenclature was defined as highlighted in Table 2. After sputter deposition, the Ca-P coatings were thermally annealed. The samples were subjected to a ramp rate of 5 °C per minute to 500 °C (from room temperature) with a soak time of 2 h and a ramp rate of 5 °C per minute back down to room temperature.
Fourier transform infrared (FTIR) spectroscopy of the samples was carried out using a BIORAD FTS 3000MX Excalibur series instrument with a PIKE diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) accessory. X-ray diffraction (XRD) of the samples was carried using a Bruker D8 Discover Diffractometer fitted with a Gobel Mirror. A Cu Kα X-ray radiation (λ = 1.540 Å) source was employed. For the grazing incidence angle XRD studies of the Ca-P coatings on the titanium coated silicon substrates the tube angle was set to 0.75°. Scans were recorded between 2Ө = 24° to 36°. X-ray photoelectron spectroscopy (XPS) of the samples were carried out using a Kratos Axis Ultra DLD spectrometer. Spectra were recorded by employing monochromated Al Kα X-rays (hν = 1486.6 electron volts (eV)). Sample charging effects on the measured BE positions were corrected by setting the lowest BE component of the C1s spectral envelope to 285.0 eV, i.e., the value generally accepted for adventitious carbon surface contamination [68]. Photoelectron spectra were further processed by subtracting a linear background and using the peak area for the most intense spectral line of each of the detected elemental species to determine the % atomic concentration. Three areas were analysed for each sample.
3 Results
3.1 Powder results
The main characteristics of the precursor powders are summarised in Tables 3 and 4. The experimental amounts of Zn and Sr in the powders, evaluated through AAS are reported as a function of the Zn and Sr content in the starting solution. It was found that not all of into Sr and Zn synthesis media added were substituted into the lattice of HAp. It implies that some of Zn and Sr ions remain in the precursor solution after precipitation. The differences between the theoretical and measured Sr and Zn concentrations might be due to Sr or Zn complexes Sr(NH3)4 2+ or Zn(NH3)4 2+ formed with the ammonia used for adjusting pH of the synthesis media [74]. Thus, Sr and Zn substitutions were inhibited, and the resulting materials were Ca-deficient. Given that the FTIR and XRD results (discussed below) did not show the presence of any by-products in the as-synthesized powders, it is most likely that the complexes were removed by washing the precipitate. Notably, the ratio between the two cations Sr/(Ca + Sr) and Zn/(Ca + Zn) in the solid products are slightly lower than those in the precursor solutions. EDX results, as reported in Table 4, shows that 10SrHAp and 10ZnHAp powders obtained by the precipitation processes have a (Ca+Sr)/P and (Ca + Zn)/P ratio < 1.67. Thus, the powders were found to be sub-stoichiometric apatite [75]. The Ca/P ratio of the HAp powder was 1.69, slightly higher than that expected for stoichiometric HAp [1, 68].
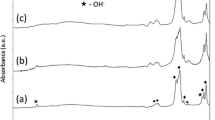
FTIR spectra of the as-synthesized HAp and Sr-, Zn-substituted HAp powders are shown in Fig. 1a–c, respectively. All of them show the characteristic absorption bands expected for the HAp powder. The absorption peaks observed at 3570 and 633 cm−1 are assigned to the vibrational modes of the [OH] group [62, 68]. However, the resolution and intensity of the absorption peak of the structural [OH] group at 633 cm−1 observed in the as-synthesized 10SrHAp and 10ZnHAp, were significantly diminished when compared to the HAp powder, highlighting the loss of OH− ions from the unit cell and the apparent substitution of Sr or Zn into the lattice of HAp [55]. The absorption band observed from 1100 to 1000 cm−1 and absorption peaks at 906, 602, 565 and 475 cm−1 are assigned to the [PO4] groups [62, 69]. The broad (weak) stretching band from 3600 to 3400 cm−1 and a bending peak at 1620 cm−1 are attributed to the adsorbed H2O molecules [60, 62, 66]. Small peaks related to [CO3] groups were detected at 1450, 1414 cm−1 and 875 cm−1 [72, 73, 76]. The peak observed at 875 cm−1 may also be associated with HPO4 2− groups. [72, 73]. However, as no other HPO4 2− were detected in these synthesised materials, the peak at 875 cm−1 is more likely a consequence of related to CO3 2− groups. The XRD results for the as-synthesized HAp and Sr-, Zn-substituted powders as shown in Fig. 2a–c, respectively. The peak positions and relative peak intensities for all of the synthesised powders correspond closely to those indicated in the International Centre for Diffraction Data (ICDD) file #01-072-1243 for HAp [77]. It was notable that all the synthesised powders exhibited significant peak broadening, however, no additional CaP phases were detected in any of the diffraction pattern obtained. In the case of the 10SrHAp substituted powder, the peak positions in the diffraction pattern are shifted to slightly lower 2-theta (2θ) values, whereas, the peak positions are relatively unchanged for the 10ZnHAp powder. The specific surface area (SSA) of precipitated powders (m2/g) was observed to increase significantly with the substitution of Ca by both Sr and Zn in the lattice, as highlighted in Table 5. This corresponds to the expected decrease in the estimated mean microcrystallite size (d BET, nm) for the substituted materials when compared to the HAp powder, as reported in Table 5. These would be in line with expectations for these materials.

FT-IR spectra of the as-synthesized a HAp, b 10SrHAp and c 10ZnHAp powders

XRD patterns of as-synthesized a HAp, b 10SrHAp and c 10ZnHAp powders
3.2 Coating results
The CaP coatings produced from the HAp, and the different 10SrHAp and 10ZnHAp target combinations were analysed using FTIR, XRD and XPS, to assess the surface properties after thermal annealing at 500 °C. The as-deposited CaP and substituted CaP coatings were not analysed in this study as previous work by the authors has shown that these surfaces are amorphous and require thermal annealing in order to produce crystalline coatings [67–69, 72, 73]. Therefore, all coatings that are described here are those that have been annealed to 500 °C only.
In the first instance, the FTIR spectrum of the coatings produced from the HAp targets were indicative of crystalline HAp, as shown in Fig. 3a. Well resolved P-O stretching vibrations were observed as expected between 1100–950 cm−1. O-P-O bending vibrations are also present between 620–560 cm−1, as shown in Table 6 [67–69]. Hydrogen phosphate bands (HPO4 2−) can also be observed at 1117 and 584 cm−1 [72, 73]. A weak O-H librational band is observed around 632 cm−1 as a very weak shoulder, with a further peak associated with O-H groups observed at 3568 cm−1. This peak may be associated with O-H stretching groups within the film [67–69]. The absence of strong OH functional groups, commonly observed at approximately 632 cm−1 and 3568 cm−1, indicate a degree of dehydroxylation within the CaP crystal structure deposited from the HAp targets under the conditions employed here. Furthermore, weak CO3 2− bands are observed between 1550–1400 cm−1 and 820–890 cm−1 [72, 76]. The peak observed at 584 cm−1 may also be due to the presence of HPO4 2− groups [72, 73]. For those coatings produced from the two 10SrHAp target configurations (SrHAp), the phosphate bands (PO4 2−) and hydrogen phosphate bands (HPO4 2−) bands are observed in largely similar peak positions as would be expected for SrHA, with shifting to slightly higher wavenumbers for most peaks observed [71–73]. Again, the OH stretching vibration at 3570 cm−1 are poorly resolved and the OH− librational band typically expected at around 632 cm−1 is absent as can be seen in Fig. 3b and Table 6. Similar results are also seen for the ZnHAp coatings as was observed for the SrHAp coatings. However, no obvious peaks for carbonate or the OH− librational vibration can be detected, as shown in Fig. 3c and Table 6. The co-substituted Sr-ZnHAp coating FTIR spectrum is again very similar to those observed for the SrHAp and ZnHAp surfaces, however, a weak shoulder indicative of OH− librational band is again present at around 631 cm−1, as shown in Fig. 3d and Table 6.

FTIR spectra for a HAp coating, b SrHAp coating, c ZnHAp coating and d Sr-ZnHAp coating (all annealed to 500 °C)
The peaks observed in the XRD patterns for the Ca-P coatings derived from the HAp targets, as shown in Fig. 4a, correspond closely to those in the ICDD file #01-072-1243 for HAp. Furthermore, the intensity of the 002 reflection at 25.9° 2θ is higher than would be expected for a HAp coating, which suggests that this coating may have a 002 preferred orientation and would be line with expectations for coatings deposited under the conditions employed here [72, 73]. The XRD pattern for the SrHAp coatings is shown in Fig. 4b, however, as expected for a Sr-substituted material, the peak positions are shifted to slightly lower 2-theta (2θ) values [71–73]. No significant peak broadening was observed for this particular coating. However, there was a notable change in the relative peak intensities of the 211 and 112 peaks with the addition of Sr to the coatings. For the ZnHAp coatings, the peak positions and intensities are very similar to those obtained for the HAp coatings produced here, with an obvious preferred 002 orientation again observed as shown in Fig. 4c. The co-substituted Sr-ZnHAp coating XRD pattern, as shown in Fig. 4d is again very similar to those observed for the SrHAp coatings, with a pronounced 002 preferred orientation and a clear difference in the relative peak intensities of the 211 and 112 peaks. In the case of all the HAp, SrHAp, ZnHAp and Sr-ZnHAp coatings, no significant amorphous background hump was observed. In addition to this no other Ca-P phases were detected in the XRD pattern for any of the coatings.

XRD patterns for a HAp coating, b SrHAp coating, c ZnHAp coating and d Sr-ZnHAp coating (all annealed to 500 °C)
The XPS results for the SrHAp, ZnHAp and Sr-ZnHAp coatings are highlighted in Fig. 5a–d, respectively and Table 7. For the HAp coating, shown in Fig. 5a, peaks corresponding to Ca2s (438.6 eV), Ca2p3/2 (347.5 eV), Ca2p1/2 (351.0 eV), P2p (133.5 eV), P2s (191.0 eV), O1s (531.5 eV), O AugerKLL (764.0 eV) and Ca AugerLMM (964.0 eV) are clearly observed and correspond closely to those reported in the literature for HAp [67–69, 72, 73]. No other elemental species were detected at least at the detection limits of the instrument (~0.1 atomic % concentration). The Ca/P ratio of the custom synthesised HAp derived coating was calculated at 1.60 ± 0.01 and is slightly lower than that expected for stoichiometric HAp, however, it is line with expectations for coatings produced under the conditions employed here [67–69]. The peak positions highlighted in Fig. 5b and in Table 7 for the SrHAp coating were all in line with those expected for Sr-substituted HAp, with clear evidence for the presence of Sr highlighted by the presence of the Sr3p3/2 at 269.5 and Sr3p1/2 at 280.1 eV [72, 73]. The Ca/P and Ca + Sr/P ratios for the SrHAp coatings were 1.38 ± 0.19 and 1.45 ± 0.22, respectively (as shown in Table 7). The spectral envelope observed at 133.5 eV consists of an overlap of the Sr3d and P2p peaks because the Sr3d 5/2 (133 ± 0.5 eV), Sr3d 3/2 (135 ± 0.5 eV) and the P2p (132–133 eV) lines are located so close together. For the ZnHAp coating, the presence of Zn in the coating is confirmed due to the presence of the Zn3p3/2 at 1022.5 and Zn3p1/2 at 1045.4 eV as shown in Fig. 5c [78]. The Ca/P and Ca+Zn/P ratios for the SrHAp coatings were 1.16 ± 0.08 and 1.20 ± 0.08, respectively (as shown in Table 7). In comparison, the wide energy survey scan for the Sr and Zn co-substituted HAp coating (Sr-ZnHAp), highlighted in Fig. 5d, shows the presence of peaks for both Sr (Sr3p3/2 at 269.4 and Sr3p1/2 at 280.1 eV) and Zn (Zn3p3/2 at 1022.4 and Zn3p1/2 at 1045.2 eV) [72, 73, 78]. The Ca/P, Ca + Sr/P, Ca + Zn/P and Ca + Sr + Zn/P ratios for the Sr-ZnHAp coatings were 1.39 ± 0.12, 1.47 ± 0.13, 1.42 ± 0.12 and 1.50 ± 0.14, respectively. If the Ca + M/Ca ratios (where M = Sr or Zn substituting ion) are also compared for the different Sr and Zn substituted coatings (as shown in Table 7), it appears that the Sr levels are comparable between the SrHAp and Sr-ZnHAp surfaces. A similar trend was observed for the Zn levels between the ZnHAp and Sr-ZnHAp surfaces. For the Sr, Zn and Sr-Zn substituted coatings, no other elemental species were detected within the detection limits of the instrument.

XPS wide energy survey scans (WESS) for a HAp coating, b SrHAp coating, c ZnHAp coating and d Sr-ZnHAp coating (all annealed to 500 °C)
4 Discussion
The present work was undertaken in order to study the deposition of Sr and Zn substituted and co-substituted CaP coatings onto titanium substrates using RF magnetron sputtering.
Before sputtering the HAp and 10SrHAp and 10ZnHAp target materials (both 10% substituted with Sr and Zn, respectively) were analysed using FTIR, XRD EDX and AAS. The purity, crystallinity and stoichiometry (Ca/P ratio of 1.69) of the HAp powder was as expected and confirmed the presence of a slightly calcium rich, carbonated HAp powder. Despite this, the XRD results highlighted that no other CaP phases were detected in the custom synthesised HAp powder. Due to the presence of the significant CO3 2− peaks between 1550–1400 cm−1 and at 875 cm−1 for the HAp powders, this indicates substitution of both the OH− and PO4 3− groups by CO3 2− (A-B type substitution) [76]. As no other significant HPO4 2− peaks were observed in the FTIR spectra for the HAp powder, it would be suggested that the peak at 875 cm−1 was due to the presence of CO3 2− in this powder sample. Therefore, the results here would suggest the HAp powder formula to be Ca10[(PO4)6−x (CO3)x][(OH)2−x (CO3) x [76]. By comparison, the purity and crystallinity of the 10SrHAp powder were largely as expected and confirmed the presence of significant levels of Sr-substitution within the HAp lattice. However, the EDX results clearly show that the 10SrHAp powder synthesised here have a (Ca+Sr)/P ratio of 1.53 (much less than the expected value of 1.67). This might be due to the generation of crystal defects from the ion substitution process [75]. The corresponding FTIR spectra highlighted that the presence of Sr in the HAp lattice resulted in significant peak broadening with respect to the PO4 3− peaks between 1200–900 cm−1, which is indicative of Sr incorporation in the HAp lattice [72, 73]. The Sr-substituted HAp powder also became more dehydroxylated as a result of the presence of Sr, with the OH- stretching and librational bands in the FTIR spectra around 3570 cm−1 and 632 cm−1 becoming much less prominent. Peak broadening and dehydroxylation are indicative of Sr incorporation of the HAp lattice. [72, 73, 79]. This usually results in structural disorder along the c-axis and it is well documented that a lack of hydroxylation within the HAp lattice affects the degree of atomic ordering in HAp [80, 81]. Small peaks related to [CO3] groups were also detected at 1450, 1414 cm−1 and 875 cm−1, which is indicative of CO3 2− groups, which are partially substituting for the PO4 3− groups in the structure of HAp (B-type carbonated HAp) [58, 76]. Despite this, no significant HPO4 2− peaks were observed in the FTIR spectra for the 10SrHAp powders as might have been expected for a Sr-substituted CaP material [72, 73]. Typically, Sr substitution for Ca in the HAp lattice results in an increase in both CO3 2− and HPO4 2− groups (especially CO3 2− groups) due to the increased lattice strain caused by the larger Sr ions [79, 82]. The XRD results for the Sr-substituted powder also highlighted that due to the incorporation of Sr into the HAp lattice, the peak positions shifted to slightly lower 2-theta (2θ) values. The peaks also exhibited significant broadening as a consequence of Sr substitution, as can be observed in Fig. 2. This would be in line with expectations and is due to substitutional strain in the lattice [83, 84]. The XRD results also highlighted that no other CaP phases were detected in any of the Sr-substituted powders. Interestingly, for the Sr-substituted HAp powder used here to produce the sputtering targets, no pronounced 002 preferred orientation was observed in the diffraction pattern. Previous studies on Sr-substituted apatites have shown that as the Sr content in HAp increases, so does c-axis orientation [71–73].
Similar observations are made for the 10ZnHAp powder when compared to that of the 10SrHAp powder, whereby the FTIR spectrum shows the powder material to be dehydroxylated (due to the diminished OH- peaks) and containing carbonate ions due to the presence of weak CO3 2− at 1450, 1414 cm−1 and 875 cm−1, which is again indicative again of B-type carbonated HAp (albeit 10ZnHAp here). The 10ZnHAp powder is also calcium deficient, with EDX giving a Ca+Zn/P ratio of only 1.51, much lower than that expected in this case (as highlighted in Table 4). The XRD results for the 10ZnHAp powders were as expected, with peaks corresponding closely to that of HAp (ICDD File# 01-072-1243). No phase impurities were detected in the 10ZnHAp powder, however, significant peak broadening was observed and a typical random crystallite orientation was observed for both the 10SrHAp and 10ZnHAp powders synthesised here and consequently used to produce the sputtering targets in this body of work. The XRD results for the 10SrHAp and 10ZnHAp powders corroborate those results highlighted from the EDX and FTIR analyses confirming that the 10SrHAp and 10ZnHAp powders contains appreciable levels of Sr and Zn, with Sr and Zn substituting for Ca in the HAp lattice. These results also suggest that the substituted powder synthesised for this study are non-stoichiometric, i.e. Ca-deficient apatite containing CO3 2− substitutions, with CO3 2− only substituting for PO4 3− (B-type carbonated HAp) [75, 76]. In both cases this would suggest a possible Ca10−X−Y−Z (M) Y (PO4)6−X−Z (CO3) Z (OH)2−X , (where M is either Sr or Zn substituting for Ca in the HAp lattice), as this allows for both a Ca/P and Sr+Ca/P lower than 1.67. Further to this, these findings also indicate increasing structural disorder or reduction in crystallite sizes as a consequence of Zn or Sr substituting for Ca in the HAp lattice. It was reported that the incorporation of a small amount of Zn into the HAp structure induces an evident reduction in the degree of crystallinity of the apatite phase, which leads to the inability of the structure to host greater amounts of Zn [85]. These observations are supported by results of BET analysis, summarized in Table 5. The SSA measurements show a significant reduction of BET particle sizes due to the incorporation of Zn into the apatite crystal structure. However, in the case of 10SrHAp powders, the results of BET analysis show an increased particle size when compared to that of 10ZnHAp. It is well documented that the Zn, Ca, and Sr ionic radii are, 0.074, 0.099, and 0.112 nm, respectively [62, 74]. The difference between the Sr and Ca ionic radii is noticeably lower as is the difference between the Zn and the Ca ionic radii. Therefore, the Zn-containing apatite is characterized by considerably higher strains and distortions of the structure [75].
After sputtering the HAp, 10SrHAp and 10ZnHAp materials onto the titanium substrates (using various different target configurations, as highlighted in Table 2) distinct changes were observed in their surfaces properties as determined using FTIR, XRD, and XPS analyses. The coatings in each case were annealed to 500 °C after deposition due to the as-deposited coatings being amorphous in nature. This is a consequence of the low sputtering power employed in this study (150 W) [67–69, 72, 73]. Others have shown that crystalline coatings can be produced by increasing the RF power [65]. However, for the purposes of this study a lower deposition power is deliberately employed as coatings produced using low power have been shown to contain only hydroxyapatite and no other additional Ca-P phases or by-products [67–69]. This would be in line with expectations of the various international standards that cover hydroxyapatite powders and coatings (ASTM 1185-03(2014), ASTM 1609—(08)2014 and ISO 13779 (Parts 2 and 3)). Furthermore, no impurities were detected on the surface of any of these coatings (as confirmed by the analyses undertaken here). The HAp derived coatings annealed to 500 °C has a Ca/P ratio of 1.60 ± 0.01, which is slightly lower than would be expected for a sputter deposited coating under the conditions employed here [67–69]. However, it should be noted that previous studies where Sr was sputter depicted onto titanium surfaces, three sputtering targets rather than two (as utilised here) were employed, and the source HAp was custom synthesised, as compared to the commercially available HAp powders used previously by the authors [68–70, 72, 73]. This may account for the slightly lower coating stoichiometry observed here for the HAp coatings. The XRD results for the same coatings produced from the two HAp targets also highlighted the expected 002 preferred c-axis orientation, as determined by XRD analyses. Typically, 002 preferred orientation in HAp results in an elevated Ca/P ratio [82]. To add to this, the FTIR results highlight that these coatings produced from the HAp precursor powders contain trace amounts of CO3 2− within the lattice, due to the presence of peaks between 1550–1400 cm−1 and 875 cm−1, as highlighted in Fig. 3a and Table 6. There is evidence for the presence of significant HPO4 2− functional groups due to the strong peaks at 1122 and 582 cm−1 (and possibly the peak at 875cm−1). Furthermore, the HAp derived coatings are also dehydroxylated. Typically, it is expected that if HAp is dehydroxylated, then the charge balance within the crystal structure must therefore be provided by other means. HAp is understood to undergo carbonate (CO3 2−) substitution within the crystal lattice at multiple sites [76, 83]. Most notably, A type substitution (for OH− groups), B type substitutions (for PO4 3− groups), or both (AB) substitution are acknowledged as providing for charge balance in the lattice structure of HAp [76, 83]. The presence of Ca10−X−Y/2[HPO4) x (PO4)]6−X−Y (CO3) Y (OH)2−X−Y species could be suggested for this coating, however, a combination of calcium deficient hydroxyapatite (CDHA) material [Ca10−X [(PO4)6–X (HPO4) X ](OH)2−X and A-B substituted materials cannot be ruled out [Ca10[(PO4)6−x (CO3) x ][(OH)2−x (CO3) x ], which may help to explain the lower Ca/P ratio obtained for this surface via XPS analysis. Typically, CDHA results in a material that has a Ca/P ratio less than 1.67 [80].
In comparison, the XPS results for the SrHAp coatings were in line with expectations for a Sr-substituted HAp coating and are similar to those reported previously [72, 73]. Peaks for Sr3p3/2 and Sr3p1/2 can be clearly observed around 269.6 and 280.1 eV in addition to the expected peaks for a CaP material [72, 73]. Figure 5b shows a representative wide energy survey spectra and high resolution spectra for the SrHAp coatings. The Ca/P ratios (and corresponding Ca + Sr/P ratio) for the same coatings were 1.38 ± 0.19 and 1.45 ± 0.22, respectively, as reported in Table 7. The Ca/P ratio reduces significantly for the SrHAp coatings when compared to the HAp surfaces, as might have been expected with the incorporation of Sr for Ca in the HAp lattice. This is corroborated by the Ca+Sr/P ratio as reported in Table 7. Furthermore, the Sr content of the coatings deposited in previous studies were lower than that of the original precursor target material, and the same trend is observed in this study [72, 73]. The corresponding FTIR spectra highlighted that for the SrHAp coating there was significant peak broadening for the PO4 3− peaks between 1200–900 cm−1. These peaks are also seen to shift to slightly lower wavenumbers and are dehydroxylated, both characteristics which are indicative of Sr substituting for Ca in the HAp lattice [72, 73]. CO3 2− peaks are also detected in the FTIR spectrum for this coating, with the peaks positions observed indicating substitution of both the OH− and PO4 3− groups by CO3 2− (A-B type substitution) [76]. Further to this, HPO4 2− bands were also clearly observed in the FTIR spectra for SrHAp coatings. It is expected that as Sr substitutes for Ca in the HAp lattice an increase in both CO3 2− and HPO4 2− groups (especially CO3 2− groups) should be observed, due to the increased lattice strain caused by the larger Sr ions as highlighted previously [86, 87]. The corresponding XRD results for SrHAp coatings highlight that they have peak positions that are shifted to slightly lower 2-theta (2θ) values with addition of Sr, as can be observed in Fig. 4. In addition the XRD results also show that the SrHAp coatings exhibit significant preferred 002 c-axis orientation, when compared to the HAp coatings and the 10SrHAp precursor powder. The intensity of the 002 peak (and therefore the 002 c-axis orientation) also appears to increase relative to the other peaks (namely the 211 peak). There is also a change in the relative peak intensities of the 211 and 112 peaks with increasing Sr content, as shown in Fig. 4b for the SrHAp surfaces. These observations would be in line with previous studies on Sr-substituted HAp, whereby it has been shown that as the Sr content in HAp increases, so does c-axis orientation [72, 73]. Considering all the results obtained here, this could suggest a formula such as Ca10−X−Y/2[HPO4) X (PO4)]6−X−Y (CO3) Y (OH)2−X−Y ) for the SrHAp coating, given the presence of both the CO3 2− and HPO4 2− groups, the apparent dehydroxylation of the coatings and the low Ca/P and C+Sr/P ratios.
In comparison, the XPS results for the ZnHAp surfaces confirmed the presence of Zn in these coatings as highlighted by WESS and Table 7. The Ca/P ratios (and corresponding Ca+Sr/P ratio) for the same coatings were 1.16 ± 0.08 and 1.20 ± 0.08, respectively, as reported in Table 7. The Ca/P ratio is significantly lower for the ZnHAp coatings when compared to both the HAp and SrHAp surfaces. The FTIR results for the ZnHAp coatings indicate that the coatings are dehydroxylated, have obvious HPO4 2− groups, but in this case, contain no real trace of any CO3 2− groups, as was observed for the HAp and SrHAp coatings. The XRD pattern obtained for this coating type is largely similar to that of HAp, with some preferred orientation observed, albeit to the lesser extent that the SrHAp surfaces. The preferred orientation observed in sputter deposited coatings, such as those deposited here, is well documented and in this case for the ZnHAp coatings the preferred orientation may be a consequence of the deposition parameters employed, as opposed to the incorporation of Zn into the HAp lattice [67–69]. No evidence for preferred orientation in Zn substituted apatites has been found in the previous literature [78, 85]. Some do report shifting of the peaks to higher 2θ values in the XRD patterns for Zn substituted materials; however, that was not observed here [88]. Typically the substitution of Zn into HAp results in a decrease of both a and c lattice parameters; however, varying points of view on this do exist in the literature [78, 85] and this is beyond the scope of this particular study. Therefore, the results here would indicate that the formula for the ZnHAp coating may be Ca10−X−Y (Zn) Y (HPO4) X (PO4)6−X (OH)2−X , given that this allows for low Ca/P and Ca+Zn/P ratios and the apparent lack of CO3 2− in the lattice. Furthermore, the apparent lack of hydroxylation, as observed in the FTIR spectrum, provide clear evidence (along with the XRD and XPS) that Zn has substituted for Ca in the ZnHAp coatings.
The results for the co-substituted Sr-ZnHAp coatings show many similarities to those obtained for both SrHAp and and ZnHAp surfaces and indicate that both Sr and Zn have been successfully substituted for Ca in the Sr-ZnHAp lattice. The reported Ca/P ratio from the XPs analyses was 1.39 ± 0.12, with Ca+Sr/P and Ca + Zn/P ratios of 1.47 ± 0.13 and 1.45 ± 0.12, respectively, as reported in Table 7. These results taken in combination with the Ca + Sr + Zn/P ratio of 1.50 ± 0.14 clearly and the WESS in Fig. 5d suggests the presence of both Sr and Zn in the co-substituted coatings. Further evidence of the successful co-substitution of the Sr-ZnHAp coatings is provided by the apparent lack of hydroxylation in the FTIR results, as highlighted in Fig. 3d and Table 6. Again, as with the Zn-HAp coatings, the co-substituted surfaces have obvious HPO4 2− groups in the FTIR spectra but no real evidence of any CO3 2− groups. The corresponding XRD pattern in Fig. 4d also shows shifting of the peaks to slightly lower 2θ values, along with strong 002 preferred orientation, which has been shown to be a consequence of Sr substituting for Ca in the lattice. This, along with the changes in the relative peak intensities of the 211 and 112 peaks in the XRD pattern, and the lack of hydroxylation and PO4 3− peak broadening from the FTIR analyses of the Sr-ZnHAp surfaces, suggest c-axis orientation and apparent disorder within the lattice of this coating. This is a direct consequence of simultaneous substitution of Sr and Zn for Ca in the lattice. As such, the suggested formula for the co-substituted Sr-ZnHAp coating produced here could be Ca10−X−Y (M) Y (HPO4) X (PO4)6−X (OH)2−X , (with M denoting both Sr and Zn ions substituting for Ca in the lattice).
If the Ca+M/Ca ratios (M is the substituting metal ion) are also taken into consideration here, the levels of Sr in the SrHAp and Sr-ZnHAp surfaces are comparable (1.05 ± 0.02 and 1.06 ± 0.01, respectively, as shown in Table 7). This is despite the difference in the number of targets used for each deposition run (two 10SrHAp targets for the SrHAp coating and one for the Sr-ZnHAp coating). A similar trend is observed for the Zn containing coatings (Ca + M/Ca), with the levels of Zn in the ZnHAp and Sr-ZnHAp surfaces reported as 1.03 ± 0.01 and 1.02 ± 0.01, respectively. By comparison, the Sr-ZnHAp coating has a Ca + Sr + Zn/Ca ratio of 1.08 ± 0.01, which clearly corroborates the successful incorporation of both Sr and Zn into the co-substituted coating. Clearly these results for the SrHAp, ZnHAp and Sr-ZnHAp coatings, highlighted from the XPS XRD and FTIR analyses confirm that they contain appreciable levels of Sr, Zn or Sr and Zn, with Sr and Zn substituting for Ca in the HAp lattice and that no other Ca-P phases or impurities (such as SrCO3), were detected for any of the substituted coatings in this study. Previous similar work undertaken by Ozeki et al. highlighted the presence of additional unwanted CaP phases after sputtering from Sr-doped HAp targets [71]. As highlighted previously, the coatings produced here were deposited using distinctly different experimental conditions, and as such it might be expected that different results were observed between the two studies. Most notably, given the relatively low sputtering power employed in this study it is less likely that all of the Sr and Zn atoms have the required energy or momentum to reach the substrate surface (in line with the Sr and Zn content of the sputtering targets. This has been described previously in the literature [71–73]. It has been suggested that the incorporation of Zn into the HAp lattice distorts the crystal lattice and hinders HAp crystal growth, and this may be inferred here from the low Ca/P and Ca + Zn/P ratios for the 10ZnHAp powder and ZnHAp coatings [78, 85]. This has been suggested to be a consequence of the mismatch between the size of the smaller Zn2+ ion (0.074nm) and that of Ca2+ (0.099 nm). Despite this, it is apparent from the results obtained here that RF magnetron sputtering can be successfully employed to sputter deposit ZnHAp, SrHAp and co-substituted Sr-ZnHAp coatings. Of particular interest here is the fact that in all cases here, the coatings produced had a distinct 002 preferred orientation (albeit more pronounced for the Sr containing coatings (SrHAp and Sr-ZnHAp). Previous work by others has shown that a preferred 002 orientation can enhance the cellular response in vitro, in particular for mesenchymal stem cells (MSC) [82]. This is most likely a consequence of the deposition parameters utilised here during sputtering, but this, with the combined therapeutic potential of Sr and Zn in the co-substituted surfaces opens up new opportunities to develop coatings that have significant in vitro and in vivo capability, beyond that already available. To date, there are very few studies that have considered the use of co-substituted Sr and Zn CaP materials in the literature [14, 62, 89], with most of the work focused towards potential co-substitutions of Mg and Sr or other ion combinations for antibacterial applications [13, 19, 23, 26, 32, 58, 61, 62, 90]. To the knowledge of the authors, this is the first successful attempt to produce Sr and Zn co-substituted HAp coatings via sputtering (and also Zn substituted HAp coatings as well). Given the obvious flexibility that sputtering provides, the development of multi-ion substituted CaP coatings is a real possibility. In the case of the co-substituted coatings produced here, previous studies have highlighted the positive effect of Sr-substitution in HAp, both in vitro and in vivo through enhancing osteoblast activity (proliferation), differentiation, and reducing osteoclast activity [7, 8, 89]. Add to this the fact that Zn has been shown to stimulate bone growth and mineralisation, lowers osteoclast activity like Sr, and has possible anti-inflammatory and antibacterial properties [7, 8, 49], the possibilities of easily delivering sputter deposited coatings with these combined properties offers significant advantages over single ion substitutions. However, detailed in vitro and in vivo studies would need to be conducted on these coatings in order to verify these findings and to study the effects of different ion concentrations when co-substituted in HAp coatings.
5 Conclusions
The aim of this particular work was to study the feasibility of using RF magnetron sputtering to deposit Sr, Zn and Sr-Zn co-substituted HAp coatings. In particular, the influence of different target configurations on the properties of the substituted sputter deposited coatings produced at a low discharge power level (150 W) were investigated. The results presented herein demonstrate that RF magnetron sputtering can be used to produce co-substituted Sr-ZnHAp coatings, and that none of the coatings produced contained any other impurity CaP phases, with XRD data showing peaks corresponding to that of ICDD file #01-072-1243 for HAp, albeit shifted to lower 2θ values due to the incorporation of Sr into the HAp lattice for Ca for the SrHAp and Sr-ZnHAp coatings. All of the coatings are calcium deficient and exhibit preferred 002 orientation, which is more pronounced for the Sr-containing surfaces. These are all properties that have been highlighted in the literature as having the potential to have a positive influence on the osteoblast response. RF magnetron sputtering therefore offers a means to control the attendant surface properties of Sr, Zn and Sr-Zn co-substituted HAp, such as the crystallinity, stoichiometry and phase purity. However, these surfaces would need to be investigated by rigorous in vitro and in vivo testing to provide further evidence of their potential to enhance osteoconduction, osteoinduction and to be osteogenic by more closely mirroring the complex chemistry of bone. This could be achieved with the potential added benefit of being anti-inflammatory and antibacterial, and delivering the next generation of biomaterials for orthopaedics.
References
Dorozhkin SV. Calcium orthophosphates: Applications in Nature, Biology, and Medicine. Singapore: Pan Stanford Publishing; 2012.
Kokubo T. Bioceramics and their clinical applications. England: Woodhead Publishing; 2008.
Ben-Nissan B, Choi AH, Roest R, Latella BA, Bendavid A. Adhesion of hydroxyapatite on titanium medical implants. In: Mucalo M, editor. Hydroxyapatite (HAp) for biomedical applications. Cambridge: Woodhead publishing series in biomaterials; 2015. pp. 21–52.
Zhang BGX, Myers DE, Wallace GG, Brandt M, Choong PFM. Bioactive coatings for orthopaedic implants—recent trends in development of implant coatings. Int J Mol Sci. 2014;15:11878–921.
Drevet R, Benhayoune H. Pulsed electrodeposition for the synthesis of strontium-substituted calcium phosphate coatings with improved dissolution properties. Mater Sci Eng C Mater Biol Appl. 2013;33:4260–5.
Lindahl C, Pujari-Palmer S, Hoess A, Ott M, Engqvist H, Xia W. The influence of Sr content in calcium phosphate coatings. Mater Sci Eng C. 2015;53:322–30.
Shepherd JH, Shepherd DV, Best SM. Substituted hydroxyapatites for bone repair. J Mater Sci: Mater Med. 2012;23:2335–47.
Mourińo V, Cattalini JP, Boccaccini AR. Metallic ions as therapeutic agents in tissue engineering scaffolds: an overview of their biological applications and strategies for new developments. J Roy Soc Interface. 2012;9:401–19.
Boanini E, Gazzano M, Bigi A. Ionic substitutions in calcium phosphates synthesized at low temperature. Acta Biomater. 2010;6:1882–94.
Stanić V, Dimitrijević S, Antić-Stanković J, Mitrić M, Jokić B, Plećaš IB, et al. Synthesis, characterization and antimicrobial activity of copper and zinc-doped hydroxyapatite nanopowders. Appl Surf Sci. 2010;256:6083–9.
Shanmugam S, Gopal B. Copper substituted hydroxyapatite and fluorapatite: synthesis, characterization and antimicrobial properties. Ceram Int. 2014;40:15655–62.
Feng QL, Cui FZ, Kim TN, Kim JW. Ag-substituted hydroxyapatite coatings with both antimicrobial effects and biocompatibility. J Mater Sci Lett. 1999;18:559–61.
Gopi D, Shinyjoy E, Kavitha L. Synthesis and spectral characterization of silver/magnesium co-substituted hydroxyapatite for biomedical applications. Spectrochim Acta—Part A Mol Biomol Spectrosc. 2014;127:286–91.
Kannan S, Goetz-Neunhoegffer F, Neubauer J, Ferreira JMF. Cosubstituition of zinc and strontium in β-triclacium phosphate: synthesis and charaterisation. Jn Amer Cer Soc. 2011;94:230–5.
Tang XL, Xiao XF, Liu RF. Structural characterization of silicon-substituted hydroxyapatite synthesized by a hydrothermal method. Mater Lett. 2005;59(29-30):3841–6.
Tian T, Jiang D, Zhang J, Lin Q. Synthesis of Si-substituted hydroxyapatite by a wet mechanochemical method. Mater Sci Eng C. 2008;28(1):57–63.
Wakamura M, Kandori K, Ishikawa T. Surface structure and composition of calcium hydroxyapatites substituted with Al(III), La(III) and Fe(III) ions. Colloids Surf A. 2000;164(2-3):297–305.
Lou W, Dong Y, Zhang H, Jin Y, Hu X, Ma J, et al. Preparation and characterization of lanthanum-incorporated hydroxyapatite coatings on titanium substrates. Int J Mol Sci. 2015;16(9):21070–86.
Zhang L, Li H, Li K, Zhang Y, Lu J, Li W. La and F co-substituted hydroxyapatite bioactive coating reinforced by SiC nanowire/ZrO2 hybrid materials for carbon/carbon composites. Ceram Int. 2016;42:2164–9.
Bianco A, Cacciotti I, Lombardi M, Montanaro L, Bemporad E, Sebastiani M. F-substituted hydroxyapatite nanopowders: thermal stability, sintering behaviour and mechanical properties. Ceram Int. 2010;36(1):313–22.
Rodriguez-Valencia C, Lopez-Alvarez M, Cochon-Cores B, Pereiro I, Serra J, Gonzalez P. Novel selnium doped hydroxyapatite coatings for biomaedical applications. J Biomed Mater Res A. 2013;101:853–61.
Kaygili O, Dorozhkin SV, Keser S. Synthesis and characterization of Ce-substituted hydroxyapatite by sol-gel method. Mater Sci Eng C. 2014;42:78–82.
Nasiri-Tabrizi B, Pingguan-Murphy B, Basirun WJ, Baradaran S. Crystallization behavior of tantalum and chlorine co-substituted hydroxyapatite nanopowders. J Ind Eng Chem. 2016;33:316–25.
Kannan S, Rebelo A, Ferreira JMF. Novel synthesis and structural characterization of fluorine and chlorine co-substituted hydroxyapatites. J Inorg Biochem. 2006;100(10):1692–7.
Sang Cho J, Um SH, Su Yoo D, Chung YC, Hye Chung S, Lee JC, et al. Enhanced osteoconductivity of sodium-substituted hydroxyapatite by system instability. J Biomed Mater Res—Part B Appl Biomater. 2014;102(5):1046–62.
Zyman Z, Tkachenko M. Sodium-carbonate co-substituted hydroxyapatite ceramics. Process Appl Ceram. 2013;7(4):153–7.
Kannan S, Ventura JMG, Ferreira JMF. Synthesis and thermal stability of potassium substituted hydroxyapatites and hydroxyapatite/β-tricalciumphosphate mixtures. Ceram Int. 2007;33(8):1489–94.
Nordström E, Karlsson K. Chemical characterization of a potassium hydroxyapatite prepared by soaking in potassium chloride and carbonate solutions. Biomed Mater Eng. 1992;2(4):185–9.
Yasukawa A, Ueda E, Kandori K, Ishikawa T. Preparation and characterization of carbonated barium-calcium hydroxyapatite solid solutions. J Colloid Interface Sci. 2005;288(2):468–74.
Hanna AA, Sherief MA, Aboelenin RM, Mousa SM. Preparation and characterizations of barium hydroxyapatite as ion exchanger. Can J Pure Appl Sci. 2010;4:1087–93.
Merry JC, Gibson IR, Best SM, Bonfield W. Synthesis and characterization of carbonate hydroxyapatite. J Mater Sci Mater Med. 1998;9(12):779–83.
Aligul B, Koseoglu NC, Aslan MH, Oral AY. Microstructural study of Mn and Si co-substituted hydroxyapatite thin films produced by a sol–gel method. Adv. Eng. Mater. 2009;11:B77–B81.
Ignjatović N, Ajduković Z, Savić V. Nanoparticles of cobalt-substituted hydroxyapatite in regeneration of mandibular osteoporotic bones. J Mater Sci Mater Med. 2013;24(2):343–54.
Swain S, Rautray TR, Narayanan R. Sr, Mg, and Co substituted hydroxyapatite coating on TiO2 nanotubes formed by electrochemical methods. Adv Sci Lett. 2016;22:482–7.
Melnikov P, Teixeira AR, Malzac A, Coelho MDB. Gallium-containing hydroxyapatite for potential use in orthopedics. Mater Chem and Phy. 2009;117:86–90.
Wakamura M. Photocatalysis by calcium hydroxyapatite modified by Ti (IV). Fujitsu Sci Tech J. 2005;41:181–90.
Hu A, Li M, Chang C, Mao D. Preparation and characterization of a titanium-substituted hydroxyapatite photocatalyst. J Mol Cat A: Chemical. 2007;267:79–85.
Wiglusz RJ, Kedziora A, Lukowiak A, Doroszkiewicz W, Strek W. Hydroxyapatite and europium (III) doped hydroxyapatite as a carrier for silver nanoparticles and their antimicrobial activity. J Biomed Nanotech. 2012;8:605–12.
Capanema NSV, Mansur AAP, Carvalho SM, Silva ARP, Ciminelli VS, Mansur HS. Niobium-doped hydroxyapatite bioceramics: synthesis, characterization and in vitro cytocompatibility. Mater. 2015;8:4191–209.
Webster TJ, Ergun C, Doremus RH, Bizios R. Hydroxyapatite with substituted magnesium, zinc, cadmium and yttrium. II Mechanisms of osteoblast adhesion. J Biomed Mater Res. 2002;59:312–7.
Terra J, Gonzalez GB, Rossi AM, Eon JG, Ellis DE. Theoretical and experimental studies of substitution of cadmium into hydroxyapatite. Phys Chem Chem Phys. 2010;21:15490–500.
Kaygili O, Dorozhkin SV, Ates T, Gursoy NC, Keser S, Yakuphanoglu F, Selçuk AB. Structural and dielectric properties of yttrium-substituted hydroxyapatites. Mater Sci Eng C Mater Biol Appl. 2015;47:333–8.
Shainburg APM, Valerio P, Zonari A, Oktar FN, Ozyegin LS, Craca MPF, Leite MF, Goes AM. Attachment and proliferation of osteoblasts on lithium-hydroxyapatite composites. Adv Mat Sci Eng. 2012;2012:10. doi:10.1155/2012/650574. Accessed 28 July 2014.
Alshemary AZ, Goh YF, Akram M, Kadir MRA, Hussain R. Microwave assisted synthesis of nano sized sulphate doped hydroxyapatite. Mater Res Bull. 2013;48:2106–10.
Ligot S, Godfroid T, Music D, Bousser E, Schneider JM, Snyders R. Tantalum-doped hydroxyapatite thin films: synthesis and characterization. Acta Materialia. 2012;60:3435–43.
Sishkumar S, Louis K, Shinyjoy E, Gopi D. Tailoring the Sm/Gd-substituted hydroxyapatite coating on biomedical AISI 316L SS: exploration of corrosion resistance, protein profiling, osteocompatibility, and osteogenic differentiation for orthopedic implant applications. Ind. Eng. Chem. Res. 2016;55:6331–44.
Webster TJ, Massa-Schlueter EA, Smith JL, Slamovich EB. Osteoblast response to hydroxyapatite doped with divalent and trivalent cations. Biomater. 2004;25:2111–21.
Kong D, Long D, Wu Y, Zhou C. Mechanical properties of hydroxyapatite-zirconia coatings prepared by magnetron sputtering. Trans. Nonferrous Met. Soc. China. 2012;22:104–10.
Cox SC, Jamshidi P, Grover LM, Mallick KK. Preparation and characterisation of nanophase Sr, Mg, and Zn substituted hydroxyapatite by aqueous precipitation. Mater Sci Eng C Mater Biol Appl. 2014;35:106–14.
Aina V, Bergandi L, Lusvardi G, Malavasi G, Imrie FE, Gibson IR, Cerrato G, Ghigo D. Sr-containing hydroxyapatite: morphologies of HAp crystals and bioactivity on osteoblast cells. Mater Sci Eng C Mater Biol Appl. 2013;33:1132–42.
Aryal S, Matsunaga K, Ching WY. Ab initio simulation of elastic and mechanical properties of Zn- and Mg-doped hydroxyapatite (HAP). J Mech Behav Biomed Mater. 2015;47:135–46.
Shepherd D. Zinc-substituted hydroxyapatite for the inhibition of osteoporosis. In: Mucalo M, editor. Hydroxyapatite (HAp) for biomedical applications. Cambridge: Woodhead publishing series in biomaterials; 2015. pp. 107–26.
Stipniece L, Salma-Ancane K, Borodajenko N, Sokolova M, Jakovlevs D, Berzina-Cimdina L. Characterization of Mg-substituted hydroxyapatite synthesized by wet chemical method. Ceram Int. 2014;40:3261–7.
Drouet C, Carayon MT, Combes C, Rey C Surface enrichment of biomimetic apatites with biologically-active ions Mg2+ and Sr2+: a preamble to the activation of bone repair materials. Mater Sci and Eng C. 2008;28:1544–50.
Ravi ND, Balu R, Kumar TTS. Strontium-substituted calcium deficient hydroxyapatite nanoparticles: synthesis, characterization, and antibacterial properties. J Am Ceram Soc. 2012;95:2700–8.
Kaygili O, Keser S, Komc M, Eroksuz Y, Dorozhkin SV, Ates T, Ozercan IH, Tatar C, Yakuphanoglu F. Strontium substituted hydroxyapatites: synthesis and determination of their structural properties, in vitro and in vivo performance. Mater Sci Eng C Mater Biol Appl. 2015;55:538–46.
Landi E, Tampieri A, Celotti G, Sprio S, Sandri M, Logroscino G. Sr-substituted hydroxyapatites for osteoporotic bone replacement. Acta Biomaterialia. 2007;3:961–9.
Kumar GS, Thamizhavel A, Yokogawa Y, Kalkura SN, Girija EK. Synthesis, characterization and in vitro studies of zinc and carbonate co-substituted nano-hydroxyapatite for biomedical applications. Mater Chem Phys. 2012;134:1127–35.
Ramya JR, Arul KT, Elayaraja K, Kalkura SN. Physicochemical and biological properties of iron and zinc ions co-doped nanocrystalline hydroxyapatite, synthesized by ultrasonication. Ceram Int. 2014;40:16707–17.
Gopi D, Ramyaa S, Rajeswaria D, Karthikeyana P, Kavitha L. Strontium, cerium co-substituted hydroxyapatite nanoparticles: synthesis, characterization, antibacterial activity towards prokaryotic strains and in vitro studies. Colloid Surface A. 2014;45:172–80.
Huang Y, Zhang X, Mao H, Li T, Zhao R, Yan Y, Pang X. Osteoblastic cell responses and antibacterial efficacy of Cu/Zn co-substituted hydroxyapatite coatings on pure titanium using electrodeposition method. RSC Adv. 2015;5:17076–86.
Kaygili O, Keser S. Sol–gel synthesis and characterization of Sr/Mg, Mg/Zn and Sr/Zn co-doped hydroxyapatites. Mater Lett. 2015;141:161–4.
Huang T, Xiao Y, Wang S, Huang Y, Liu X, Wu F, Gu Z. Nanostructured Si, Mg, CO3 2− substituted hydroxyapatite coatings deposited by liquid precursor plasma spraying: synthesis and characterization. J Thermal Spray Tech. 2011;20:829–36.
O’Sullivan C, O’Hare P, O’Leary ND, Crean AM, Ryan K, Dobson ADW, O’Neill L. Deposition of substituted apatites with anticolonizing properties onto titanium surfaces using a novel blasting process. J. Biomed. Mat. Res B: Appl. Biomater. 2010;95B:141–9.
van Dijk K, Schaeken HG, Wolke JGC, Maree CHM, Habraken FHPM, Verhoeven J, Jansen JA. Influence of discharge power level on the properties of hydroxyapatite films deposited on Ti6A14V with RF magnetron sputtering. J Biomed Mat Res. 1995;29:269–76.
Yonggang Y, Wolke JGC, Yubao L, Jansen JA. The influence of discharge power and heat treatment on calcium phosphate coatings prepared by RF magnetron sputtering deposition. J Mater Sci: Mater Med. 2007;18:1061–9.
Boyd AR, Akay M, Meenan BJ. Influence of target surface degradation on the properties of RF magnetron‐sputtered calcium phosphate coatings. Surf Inter Anal. 2003;35:188–98.
Boyd AR, Meenan BJ, Leyland NS. Surface characterisation of the evolving nature of radio frequency (RF) magnetron sputter deposited calcium phosphate thin films after exposure to physiological solution. Surf Coat Tech. 2006;200:6002–13.
Boyd AR, O’Kane C, Meenan BJ. Control of calcium phosphate thin film stoichiometry using multi-target sputter deposition. Surf Coat Tech. 2013;233:131–9.
Thian ES, Huang J, Best SM, Barber ZH, Bonfield W. Surface modification of magnetron-sputtered hydroxyapatite thin films via silicon substitution for orthopaedic and dental applications. Surf Coat Technol. 2011;205:3472–7.
Ozeki K, Hoshino T, Aoki H, Masuzawa T. Phase composition of sputtered film from a mixture of hydroxyapatite and strontium-apatite. J Mater Sci Technol. 2013;29:1–6.
Boyd A, Randolph LD, Rutledge L, Meenan BJ. Strontium-substituted hydroxyapatite coatings deposited via a co-deposition sputter technique. Mater Sci & Eng C. 2015;46:290–300.
Boyd A, Rutledge L, Randolph LD, Mutreja I, Meenan BJ. The deposition of strontium-substituted hydroxyapatite coatings. J Mater Sci: Mater Med. 2015;26:1–14.
Li MO, Xiao XF, Liu RF, Chen CY, Huang LZ. Structural characterization of zinc-substituted hydroxyapatite prepared by hydrothermal method. J Mater Sci: Mater Med. 2008;19(2):797–803.
Quilitz M, Steingröver K, Veith M. Effect of the Ca/P ratio on the dielectric properties of nanoscaled substoichiometric hydroxyapatite. J Mater Sci: Mater Med. 2010;21:399–405.
Gibson IR, Bonfield W. Novel synthesis and characterization of an AB-type carbonate-substituted hydroxyapatite. J Biomed Mater Res. 2002;59:697–708.
Fadeeva IV, Bakunova NV, Komlev VS, Medvecký L, Fomin AS, Gurin AN, Barinov SM. Zinc- and silver- substituted hydroxyapatite: synthesis and properties. Doklady Akademii Nauk. 2012;442:780–3.
Thian ES, Konishi T, Kawanobe Y, Lim PN, Choong C, Ho B, Aizawa M. Zinc-substituted hydroxyapatite: a biomaterial with enhanced bioactivity and antibacterial properties. J Mater Sci: Mater Med. 2013;24:437–45.
Yu T, Ye J, Wang Y. Preparation and characterization of a novel strontium-containing calcium phosphate cement with the two-step hydration process. Acta Biomater. 2009;5:2717–27.
Dorozhkin SV. Bioceramics of calcium orthophosphates. Biomater. 2010;31:1465–85.
Yang Y, Perez-Amodio S, Barre‘ re-de Groot FYF, Everts V, van Blitterswijk CA, Habibovic P. The effects of inorganic additives to calcium phosphate on in vitro behaviour of osteoblasts and osteoclasts. Biomater. 2010;31:2976–89.
Kim H, Camata RP, Chowdhury S, Vohra YK. In vitro dissolution and mechanical behavior of c-axis preferentially oriented hydroxyapatite thin films fabricated by pulsed laser deposition. Acta Biomater. 2010;6:3234–41.
Wopenka B, Pasteris JD. A mineralogical perspective on the apatite in bone. Mat Sci Eng C. 2005;25:131–43.
Suganthi RV, Elayaraja K, Joshy MIA, Chandra VS, Girija EK, Kalkura SN. Fibrous growth of strontium substituted hydroxyapatite and its drug release. Mat Sci Eng C. 2010;31:593–9.
Ren F, Xin R, Ge X, Leng Y. Characterization and structural analysis of zinc-substituted hydroxyapatites. Acta Biomaterialia. 2009;5:3141–9.
Vignoles M, Bone G, Young RA. Occurrence of nitrogenous species in precipitated B-type carbonated hydroxyapatites. Calcif Tissue Int. 1987;40:64–70.
Kavitha M, Subramanian R, Narayanan R, Udhayabanu V. Solution combustion synthesis and characterization of strontium substituted hydroxyapatite nanocrystals. Powd Tech. 2014;253:129–37.
Norhidayu D, Sopyan I, Ramesh S. Development of zinc doped hydroxyapatite for bone implant applications. ICCBT. 2008;F-24:257–70.
Zreiqat H, Ramaswamy Y, Wu C, Paschalidis A, Lu Z, James B, Birke O, McDonald M, Little D, Dunstan CR. The incorporation of strontium and zinc into a calcium-silicon ceramic for bone tissue engineering. Biomater. 2010;31:3175–84.
Imrie FE, Aina V, Lusvardi G, Malavasi G, Gibson IR, Cerrato G, Annaz B. Synthesis and characterisation of strontium and magnesium co-substituted biphasic calcium phosphates. Key Eng Mater. 2013;529-30:88–93.
Acknowledgements
This work has been supported by the National Research Programme No. 2014.10-4/VPP-3/21 “Multifunctional materials and composites, photonics and nanotechnology (IMIS2)” Project No. 4 “Nanomaterials and nanotechnologies for medical applications”. The authors would also like to acknowledge the PhD funding provided by of the Department of Employment and Learning (Northern Ireland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Rights and permissions
About this article

Cite this article
Robinson, L., Salma-Ancane, K., Stipniece, L. et al. The deposition of strontium and zinc Co-substituted hydroxyapatite coatings. J Mater Sci: Mater Med 28, 51 (2017). https://doi.org/10.1007/s10856-017-5846-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-017-5846-2