
Abstract
In this study, chitosan–Laponite nanocomposite coatings with bone regenerative potential and controlled drug-release capacity are prepared by electrophoretic deposition technique. The controlled release of a glycopeptide drug, i.e. vancomycin, is attained by the intercalation of the polymer and drug macromolecules into silicate galleries. Fourier-transform infrared spectrometry reveals electrostatic interactions between the charged structure of clay and the amine and hydroxyl groups of chitosan and vancomycin, leading to a complex positively-charged system with high electrophoretic mobility. By applying electric field the charged particles are deposited on the surface of titanium foils and uniform chitosan films containing 25–55 wt% Laponite and 937–1655 µg/cm2 vancomycin are obtained. Nanocomposite films exhibit improved cell attachment with higher cell viability. Alkaline phosphatase assay reveals enhanced cell proliferation due the gradual dissolution of Laponite particles into the culture medium. In-vitro drug-release studies show lower release rate through a longer period for the nanocomposite compared to pristine chitosan.
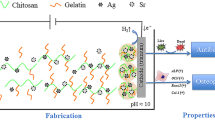
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Recently, biopolymer-clay mineral nanocomposites have attracted considerable research interest in biomedical and pharmaceutical applications [1]. The superior properties of these materials such as biodegradability, biocompatibility and tunable mechanical properties are essential keys for tissue engineering and controlled drug delivery [1, 2]. Among biopolymers, chitosan (CS), a positively charged semi-crystalline polysaccharide, has been widely used to prepare nanocomposites with many different types of clay [3, 4]. Chitosan is mainly composed of N-acetyl d-glucosamine and d-glucosamine units [5]. The antibacterial nature [6, 7], biocompatibility [8], biodegradability [9] and non-toxicity [10] combined with some of the polymeric properties of chitosan have made this polysaccharide as a promising candidate for tissue engineering [5, 11] and drug delivery applications [12]. The biocompatibility of chitosan is originated from its structural similarity to glycosaminoglycans, which are the major component of the extracellular matrix (ECM) of bone and cartilage [13]. Chitosan has also widely been used as an excipient, e.g. as film forming or gelling agent in the field of pharmaceutics [14]. Due to its controlled-release properties, chitosan is commonly used in the preparation of tablets, beads, microspheres, hydrogels and films [12]. Recent studies have shown that not only clay minerals enhance the thermal stability and mechanical properties of chitosan [4], but also improve the drug encapsulation and release rate [15]. As a result, clay minerals such as Montmorillonite [16], Laponite [17] and magnesium aluminum silicate [14] are used in pharmaceutical technology as active agents for the development of new hybrid drug delivery systems with suitable biological activities. This is primary due to their desirable properties including chemical inertness, biocompatibility, high specific surfaces areas, large pore volumes, and mechanical stability [18].
Laponite (LP; \({\text{Na}}_{0.7}^{ + } [\left( {{\text{Mg}}_{5.5} {\text{Li}}_{0.5} } \right){\text{Si}}_{8} {\text{O}}_{20} \left( {\text{OH}} \right)_{4} ]_{0.7}^{ - } )\), is of particular interest for bone tissue engineering as it has a similar chemical composition to the bioactive glasses [19, 20]. This nano-layered clay is biocompatible and non-toxic [21] with high potential to enhance osteoblast proliferation and cell differentiation [20]. Therefore, Laponite is considered as the next generation of bioactive materials, not only because of its unique biological properties but also due to its advantages such as high-purity, low cost and easy processing [22]. Therefore, chitosan–Laponite (CS–LP) nanocomposites are potential polymeric nanocomposites of interest for biomedical applications including tissue engineering, controlled drug delivery, and biosensors. Notably, negatively charged Laponite particles can form biodegradable nanocomposites with positively charged chitosan through strong electrostatic interaction [1]. Recently, CS–LP nanocomposites have successfully been utilized to prepare a new type of an amperometric biosensor for phenol determination [19] and amperometric glucose biosensor [23]. Yang et al. [24] prepared CS–LP hydrogel beads as a drug delivery carrier and showed that the incorporation of Laponite significantly improved the swelling behavior, enhanced the drug entrapment efficiency, and slowed down the drug release behavior in contrast to the pure chitosan beads.
The aim of the present work is to prepare CS–LP nanocomposite films with antibiotic-eluting capacity on titanium implants. These films can potentially be used for reducing of microbial risk of bone implants as this infection is the major issue in orthopaedic surgery [25]. Prophylactic systematic antibiotic therapies, which are administered routinely to patients who receive an orthopaedic device, usually fail because of the poor penetration into bone and possible systemic or organ-specific toxicity [26]. Therefore, local administration of antibiotics from implants through antibacterial drug-eluting coatings has received much attention in recent years [27]. We adopted the facile and reproducible electrophoretic method to prepare CS–LP nanocomposite coatings on titanium implants with antibiotic-eluting potential and bone regeneration capacity. To the best of our knowledge, this study is the first to apply electrophoretic deposition to the synthesis of biodegradable drug-eluting CS–LP nanocomposite coatings. The advantages of the electrophoretic deposition process include low-cost, high versatility, simplicity, processing of complex shapes, high purity and microstructural homogeneity of the deposits [28, 29]. This technique has been used for preparation of chitosan [30, 31] and chitosan-based composite coatings [29, 32]. Recently, this technique has been applied to prepare drug-eluting chitosan [33], chitosan–bioactive glass [34], and chitosan–graphene oxide composite coatings [35]. In chitosan/antibiotic coatings, rapid destruction of hydrogen bonding between the drug and chitosan by polar water molecules and rapid degradation/de-attachment of the coating lead to a non-uniform and short releasing behavior [33]. Incorporation of bioactive glass particles prolongs the release time, however, the amount of initial burst release is intensified [34]. Graphene oxide makes the release rate more controllable but reduces osteogenesis [35]. Motivated by our previous studies, in this paper, we present the effect of Laponite clay on the physical, chemical, and biological properties of drug-eluting chitosan-based coatings deposited on the surface of titanium foils. By studying the drug release profile versus time (up to 28 days) and in vitro cytocompatibility using MG-63 osteoblast-like cell lines, we introduce a new strategy for preparing antibacterial drug-eluting biocomposite coatings for bone implants.
2 Methods
2.1 Electrophoretic deposition
An aqueous chitosan solution with a concentration of 0.5 g/l was prepared by dissolving chitosan flakes (85 % deacetylated, 190–310 kDa; Sigma-Aldrich, USA) in 1 % (v/v) glacial acetic acid (Merck, Germany). The pH of the solution was adjusted to a value of 3 ± 0.05 by a Metrohm® 827 pH Lab Meter (USA Inc.). The solution was stirred overnight and then filtered to remove residuals (Whatman® quantitative filter paper, Grade 42, pore size 2.5 µm). Laponite RD, a synthetic layered silicate consisting of nanoparticles (diameter, 25–30 nm; thickness, 1 nm), was obtained from Southern Clay, USA. Suspensions containing different amounts of Laponite (0.05, 0.1, 0.25, 0.5 and 1 g/l) were prepared by dispersing Laponite particles in chitosan solution using magnetic stirring. To prepare drug-eluting coatings, vancomycin, which is a glycopeptide antibiotic with high activity against Gram-positive bacteria [36], was utilized. This antibiotic has frequently been utilized to treat and prevent osteomyelitis and deep infections [37]. Based on our previous studies [33, 34], the drug-loaded coatings were prepared by adding 2 g/l vancomycin (Dana Tabriz Company, Iran) to the chitosan-based suspensions followed by magnetic stirring for 30 min. Titanium foils (ASTM B265, ATI Allegheny Ludlum, USA) with dimensions of 10 × 20 × 0.45 mm3 were utilized as both deposition substrate and counter-electrode in the cell. The electrodes were mounted parallel to each other with the separation of 10 mm in a glass container containing 25 ml of the suspensions. A dc voltage of 10 V was applied and remained on for 10 min. After deposition, the cathodic films were stored and dried in a desiccator for further characterizations.
2.2 Materials characterization
In order to study the electrophoretic potential of different suspensions, zeta potential measurements were carried out by a Malvern Zeta Sizer (Model HS C1330-3000, UK). After deposition, the thickness and microstructural features of the films were studied by scanning electron microscopy (SEM, VEGA TESCAN, Czech Republic). The surface roughness was determined by employing a DME DualScope™ C-26 Atomic force microscope (AFM) in a non-contact mode. Surface hydrophilicity was determined by an OCA15 plus video-based optical contact angle meter (Dataphysics Instruments GmbH, Filderstadt, Germany). The images of a water droplet (4 µl) spreading on the sample surface were recorded by a camera, and then analyzed using the software supplied by the manufacturer. In order to identify the chemical structure of coatings and possible interaction between the composite components, Fourier transform infrared spectroscopy (FTIR, ABB Bomem MB100, USA) in the range of 400–4000 cm−1 with a resolution of 4 cm−1 was utilized. Small-angle X-ray diffraction patterns of the samples were obtained using a XRD, X’Pert Pro MPD, PANalytical diffractometer with CuKα radiation at 40 kV in the scan range of 2θ from 1° to 10° at a scan rate of 1 min−1. The d-spacing was calculated by Bragg’s formula where k = 0.154 nm. In order to estimate the composition of the coatings and changes in the thermal stability of chitosan films, thermo-gravimetric analysis (TGA/DSC 1, METTLER TOLEDO, Switzerland) under a nitrogen atmosphere was performed. A heating rate of 10 °C min−1 was utilized. Before doing FTIR, XRD and TGA, the coatings were removed from the substrates.
2.3 Drug release studies
The release profile of the drug from the coatings was determined by immersing the specimens in the phosphate-buffered saline solution (PBS) at controlled pH and temperature of 7.4 and 37 °C. The amount of released drug was monitored by an ultraviolent-visible spectrophotometer (PerkinElmer, Lambda 25, UV/VIS Spectrophotometer, USA). A series of standard vancomycin solutions in PBS (5–100 µg/ml) were prepared to obtain a linear calibration curve (R2 = 0.99) that obeys Beer’s law. A UV standard absorbance curve for vancomycin at 280 nm was then established. To eliminate any possible interference of the degraded products, blank solutions for the UV/VIS spectroscopy assay was prepared by collecting solutions from the coatings free of drug at the same incubation time as the drug-eluting period. All release studies were carried out in triplicate. The accumulated drug in the solution was calculated and plotted versus time to determine the drug release kinetics. The results are presented in terms of cumulative release as a function of time.
where Mt is the cumulative amount of released drug from the coatings at time t, and Mo the initial amount of loaded drug.
2.4 In vitro cell-material interactions
Human osteosarcoma cells (MG-63 osteoblast-like) were obtained from the National Cell Bank, Iran Pasteur Institute (Tehran, Iran). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, USA) with 10 % Fetal Bovine Serum (FBS, Sigma-Aldrich, USA) at 37 °C in a humidified atmosphere with 5 % CO2. Prior to cell seeding, the samples were washed with an aqueous solution containing ethanol to remove excess acetic acid followed by UV-sterilization for 40 min. The studied biological properties included cell attachment, cytotoxicity and alkaline phosphates (ALP) activity as described below. For each experiment, three replicates were used per group. Tissue culture polystyrene (TPS) was used as a negative control groups for all tests.
2.4.1 Cell viability
The standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) protocol was utilized to assay the cell viability. Briefly, 2 × 105 cells were seeded on the specimens within a 12-well plate and were incubated at 37 °C in 5 % CO2 for 1, 5, and 10 days. After each interval, MTT solution at a concentration of 5 mg/ml was added to each well and the cells were incubated for another 4 h. Upon removal of the MTT solution, the formed formazan crystals were solubilized with isopropanol for 15 min and the amount of produced formazan (that was proportional to the number of viable cells) was determined by an ELISA reader (BioTekmicroplate reader, USA) at 570 nm.
2.4.2 Cell attachment
In order to evaluate the cell attachment on the surface of the electrodeposited films, 2 × 105 MG-63 osteoblast-like cells were added on the surface of the specimens and incubated in DMEM supplemented with 10 % FBS at 37 °C for 1, 5 and 10 days. Afterward, the DMEM was removed and cells were incubated in 100 μl/well Neutral Red (NR) solution for 4 h. The stain extraction solution (1 % glacial acetic acid, 50 % ethanol, and 49 % distilled water) was then replaced with NR solution in order to dissolve deposited particles and was re-incubated in 37 °C for 15 min. The absorbance was measured at 570 nm using the ELISA reader.
2.4.3 Alkaline phosphates activity
The samples were cultured in the cell medium as identical to the MTT protocol. To examine differentiation of the cells at different time intervals (1, 5, and 10 days), 20 μl of cell supernatant was added to 1000 μl of ALP’s kit (Pars Azmun, Iran) according to the manufacturer’s protocol at 37 °C. The absorbance was read at 405 nm using a NanoDrop 2000c Spectrophotometer, USA.
2.4.4 Cell morphology
The cultured cells on the surface of the films were fixed with Karnovsky (2.5 % glutaraldehyde, 4 % paraformaldehyde) solution for 90 min. After dehydration in a graded series of ethanol, the samples were vacuum dried and gold coated for SEM examinations by a VEGA TESCAN SEM (Czech Republic).
2.5 Statistical analysis
The experiments were performed in triple numbers per series and the results were expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) with P < 0.05 as the level of significance was utilized for statistical analysis.
3 Results
3.1 Characterizations of electrodeposited films
Various analytical techniques were utilized to characterize the drug-eluting CS-LP nanocomposite coatings prepared by EPD. Figure 1 shows SEM images of the coatings deposited on the surface of titanium foils. The pristine chitosan film exhibits a homogenous and smooth surface morphology (Fig. 1a). The CS-LP film has a rough morphology containing particulate structure embedded in the chitosan matrix (Fig. 1b). Further study indicates a homogeneous distribution of spherical LP particles in the polymeric matrix, although slight agglomeration is also seen (Fig. 1c). The microstructure of drug-eluting nanocomposite coating consists of a chitosan matrix with micron-sized LP particles and drug (VAN) clusters (Fig. 1d). A magnified image of the particle clusters is shown in Fig. 1e, and a cross-sectional SEM image of the drug-eluting film is seen in Fig. 1f. The film thickness is about 70 µm. As reported elsewhere [35], the pristine CS films electrodeposited at the same condition have a thickness of about 40 µm. Thus, the film thickness increased approximately 75 % by adding LP and VAN particles.

Surface morphology of chitosan-based coatings: a CS; b, c CS-LP (43 wt%); d, e CS-LP (43 wt%)-VAN. f Cross-sectional SEM image of the drug-eluting composite coating on Ti substrate. The Laponite particles (white arrows) and Laponite–VAN clusters (red arrows) are indicated
The approximate composition and changes in thermal stability of the composite coatings were investigated by using TGA. The films were scratched off from the Ti substrate and heated up to 800 °C in the nitrogen atmosphere. Figure 2 shows the thermal degradation of CS coatings containing different amounts of LP and their first order derivatives with respect to temperature (DTG). The TGA pattern indicates a three-step weight loss. The first two steps (up to ~220 °C) can be attributed to the liberation of adsorbed water. The third step (mainly around 270 °C) is attributed to the thermal decomposition of the polymer. Although the pattern of all films is rather similar, the temperature at which thermal degradation starts (as a criterion of the thermal stability) is varied by the LP contents. For instance, the degradation deep of CS is shifted from 267 to 274 °C and 282 °C with the addition of 0.5 and 1 g/l LP in the EPD suspensions (Fig. 2b). This observation can be an indicator of interactions of the polymer chains with the clay particles, i.e. formation of intercalated nanocomposite through penetration of CS chains into the galleries of the LP particles [38]. Under this circumstance, the thermal motion of chitosan macromolecules is restricted by LP plates [38], which enhances thermal stability of the composite coatings compared to the pristine CS. The TGA results also indicate different weight losses for CS and CS–LP composites over the entire thermal treatment. From this data, one is able to estimate the LP amount embedded in the CS matrix, as shown in Fig. 2c. An increasing trend in the LP amount in the composite film with the EPD composition is seen; however, this relationship does not follow the Hamaker’s equation (the linear trend) [39]. The reason could be attributed to interactions between polysaccharide and clay particles at nano- and macro- molecular levels and agglomeration of the particles in the EPD suspension at high concentrations.

a, b Thermogravimetric traces of electrodeposited coatings at a heating rate of 10 °C per min in nitrogen atmosphere. c Concentration of LP clay in the composite coating after EPD of composite suspensions with varying LP content
To further analysis such interactions, XRD and FITR were employed (Fig. 3). The XRD pattern of LP clay shows a characteristic diffraction peak at 2θ = 6.57° corresponding to (001) plane with d-spacing of 13.43 Å. This characteristic peak is shifted to a lower angle of 3.79° for the CS-LP (43 wt%) nanocomposite film, which means increasing of crystallographic plan spacing of LP particles by the polymer. No characteristic peak for the Laponite nanoparticles could be detectable in the XRD pattern of drug-eluting CS–LP nanocomposite films, which demonstrates good exfoliation of Laponite nanoparticles in chitosan matrix. These findings can further be supported by FTIR analysis (Fig. 3b). The FTIR spectrum of CS consists of a broad band at ~3450 cm−1 (the stretching vibration of N–H and hydroxyl group), peaks at 2900 and 2875 cm−1 (typical C-H stretch vibrations), those at 1643, 1580 and 1320 cm−1 (amides I, II and III, respectively), and peaks at 1154, 1078 and 1030 cm−1 (C–O stretching vibrations) [30]. The main characteristic peaks of pristine LP are at 3440, 1640, 1004 and 655 cm−1, which correspond to H–OH stretching, H–OH bending adsorption, Si–O stretching and Mg–O band, respectively [19]. In the CS–LP (43 wt%) nanocomposite film, however, shifts in the IR bands of amines to lower wavenumbers (i.e., 1643–1634 cm−1 and 1580–1565 cm−1) are seen. Therefore, electrostatic interactions between the negatively charged structure of clay and the amine groups of chitosan may happen. Similarly, such shifts in the amine and hydroxyl bands are detectable in the presence of the drug molecules, revealing intercalations of CS and VAN with the LP particles through interlayer penetration and/or surface adsorption [40].

a XRD and b FTIR spectra of CS-LP (43 wt%) nanocomposite coatings containing VAN compared to the non-eluting coating. AFM images show the surface topography of c CS and d CS-LP (43 wt%)-VAN coatings
Since the surface topography [41] and hydrophilicity [42] of the coatings influence their biocompatibility and cellular behavior, AFM and contact angle measurement were performed. The topographic surfaces of CS and drug-eluting CS–LP films are shown in Fig. 3c, d. It is seen that the addition of LP particles and the drug increases the surface roughness of the CS films. Contact angle measurements also show that these additives enhance the hydrophilicity of CS (see Electronic Supplementary Information (ESI) S1). This improvement depends on the concentration of LP and VAN as these particles are more hydrophilic than CS.
3.2 Drug release behaviour
A fixed amount of vancomycin (2 g/l) was added to the EPD suspensions of CS containing different amounts of LP particles (up to 1 g/l). The amount of loaded drug in the composite coatings was estimated by long-term immersion of the films in PBS solution. The amount of incorporated drug was increased from 228 µg/cm2 for the CS to 1090 µg/cm2 for the CS–LP composite (1:1 weight ratio in the EPD suspension). This result indicates the positive effect of LP particles on the drug encapsulation efficiency of chitosan-based coatings. Figure 4a shows a typical release profile of the electrodeposited films for CS–VAN and CS–LP (43 wt%)-VAN systems. An initially burst release (IBR) followed by a reduced release rate is visible. The amount of IBR depends on the concentration of LP clay, as shown in Fig. 4b. Since IBR is related to rapid dissolution of the drugs located at or close to the surface of the coatings or at the pore walls of chitosan hydrogel [34, 40], the lower values of IBR in the presence of LP particles indicate the effect of drug-clay interactions on the release profile. These interactions also influence the subsequent release rate. As seen in Fig. 4a, the composite films exhibit a remarkable slower release rate. After the IBR period (about 1 h), the accumulative released concentration follows a linear trend with respect to the square root of incubation time in PBS solution (Fig. 4c). This behavior indicates a diffusion-controlled mechanism for the release [43] as is discussed in the next section.

Cumulative drug release of the chitosan-based coatings versus immersion time; b the amount of burst release from chitosan-based coatings; c cumulative drug release of the chitosan-based coatings versus square root of immersion time in PBS at 37 °C
3.3 In vitro cell culture studies
The biocompatibility of electrodeposited coatings and their interactions with MG-63 osteoblast-like cells were evaluated by in vitro cell culture experiments. Figure 5a shows the effect of the material system on the cell viability, as quantified by means of MTT assay. At a short incubation time (1 day), higher viability is attained for the electrodeposited films compared to TPS, particularly for the CS–LP (43 wt%) coating. The viability of the films gradually decreases over time, but it is still high enough to be considered the coatings biocompatible (>90 % after 10 days). This decline in the viability may be due to cell proliferation, resulting in cells occupying all the available space on the films (geometrical restriction) [44]. The results also highlight the effect of LP particles on the improved cytocompatibility of the nanocomposite films compared with the CS coatings. Figure 5b shows the results of ALP assay. Alkaline phosphatase activity is commonly used as an early marker for the osteoblast phenotype [45]. It appears that, except CS film incubated for 5 days, the ALP activity increases with the time. The higher ALP activity of the nanocomposite film than that of the CS coating is also noticeable. Therefore, it appears that LP particles promote proliferation and cell viability of CS. Similar results have been observed for alginate-Laponite nanocomposites [21]. In the meantime, the incorporation of drug in the nanocomposite reduces the cell viability and the ALP activity. This behavior can be attributed to the drug release in the culture medium. Figure 5c shows the number of osteoblast cells adhered on the surface of the coatings at different time intervals. Typical SEM images of cells adhered to the surface are shown in Fig. 5d, e. As seen, the cell density on the surface of the nanocomposite coating is notably higher than that of pristine CS film. From the morphological point of view, the adhered cell on the surface of CS film has a rounded morphology with little evidence of interactions with the surrounding matrix. In contrast, a healthy spindle-shape cell which is spread well on the surface is seen for the nanocomposite. The presence of multiple filopodia extended from the cell to the substrate provides strong evidence that the cells are well attached to the nanocomposite coating [46].

Cellular behaviour of the prepared coatings: a MTT viability; b ALP activities and c cell attachments. SEM images of MG-63 osteoblast-like cells cultured on the surface of d CS and e CS-LP (43 wt%)-VAN coatings for 4 days. * denotes significant difference between TPS and EPD coatings (P < 0.05). # denotes significant difference between CS and composite coatings (P < 0.05)
4 Discussion
The presented results demonstrated the suitability of EPD as a processing method to prepare CS–LP composite films with controlled drug-release and bone regenerative capacity. The mechanisms of electrophoretic deposition of CS and CS–VAN coatings have been previously studied [33, 47]. Chitosan electrophoresis toward the cathode occurs due to protonation of its amino groups at acidic pH and it deposits at the cathode due to water electrolysis and consequent increase in the local pH (above pKa of chitosan). Zeta potential measurement at pH 3 yielded values of 56.8 ± 1.6 and 57.7 ± 0.9 mV for CS–LP suspensions without and with drug, respectively. Both suspensions have good positive values of zeta potential; therefore, cathodic electrodeposition is feasible. In the acidic solution, CS and VAN macromolecules become positively charged by protonation, and easily co-deposited on the surface of the negative electrode. In the composite suspension, the LP particles, which are initially negatively charged [19], are intercalated with the polymer macromolecules through cation exchange. This process occurs when cations within the clay (Na+) are exchanged for cations of polymeric solution. In other words, the positively charged polymers interact with the negatively charged silicate surfaces [19, 44] or intercalate them via an ion exchange reaction [48], as supported by XRD and FTIR (Fig. 3). By applying electric field, the positively charged complexes move toward the cathode and co-deposition occurs.
The prepared nanocomposite coatings can potentially improve the bactericidal and osseointegration of Ti orthopaedic implants. The drug release study indicated the effect of LP particles on the vancomycin encapsulation efficiency of the chitosan-based coatings (Fig. 4). As explained above, intercalation and interactions of the clay particles with the drug molecules enhanced the co-electrophoretic deposition of the multi-component system. During the immersion in PBS, the drug is released by a two-stage mechanism including an initial burst release followed by a linear release with the square root of time (a diffusion controlled process). Degradation/de-attachment of chitosan coatings and diffusion of water through the hydrogel matrix are two mechanisms that affect the release of vancomycin [24, 33].
The slower release rate of drug from the CS–LP composite coatings compared to the pristine CS is attributed to the interactions between LP layers with the polymer matrix as well as physical (geometrical) barrier against diffusion. The interactions between silicate layers with chitosan and vancomycin macromolecules could be Columbic interactions, van der Waals force, and H-bonding [44]. Yuan et al. [40] showed that the cross-linked chitosan exhibited controlled swelling behavior because of the dense macromolecular chains. The addition of clay to chitosan builds a strong cross-linking structure because of the negatively charged clay and positively charged NH3 + groups of chitosan. The formation of this structure influences the swelling behavior of the nanocomposite and consequently affects diffusion of the drug through the bulk entity. Based on the experimental results, it is suggestible that electrostatic interactions between the positively charged polymers and negatively charged sites on the clay are responsible for the lower release rate of drug in the composite coating. In fact, ionic interactions between positively charged CS and negatively charged LP strengthen the polymer network by acting as additional cross-linkers. It reduces the overall hydration and swelling of the nanocomposite [45], consequently decreases the dissolution/degradation rate of the nanocomposites. Therefore, while the degradation/detachment of the coating is the main release mechanism for CS, diffusion of entities through the composite coating becomes operative.
The biological tests performed in the present work have shown the effect of LP clay on the biocompatibility, adhesion and differentiation of osteoblast-like cells. It is known that the biocompatibility of orthopaedic implants is affected by their surface characteristics such as hydrophilicity, topography, roughness and chemical composition [41, 49]. AFM and contact angle measurement showed that the composite coatings had rougher surfaces with enhanced wettability. In addition to roughness and wettability enhancement, dissolution of LP particles into PBS solution (see EIS S2) and their ionic in the culture medium could affect the interaction of cells with the material. The effect of LP particles on the cellular responses of polymer-matrix composites has been demonstrated in many studies [21, 22]. Gaharwar et al. [22] have illustrated that LP particles do not show acute toxicity to preosteoblast cells at low concentrations, and thus can potentially be used for various biomedical applications. The results of MTT assay and ALP activity showed that cytocompatibility and proliferation of MG-63 cells on the CS–LP coating were higher than that of the pristine CS (Fig. 5). The effect of LP clay on the bioactivity of the CS coatings can be explained by its gradual dissolution in the medium and its influence on the adsorption of adhesion proteins. Proteins, including adhesion proteins such as vitronectin and fibronectin, can easily be adsorbed onto the surface of silicate nanoparticles, as the nanoparticles can provide focal points for cell adhesion. On the other hand, silica up-regulates the expression of genes that are involved in osteoblast proliferation and differentiation [50].
5 Conclusion
In summary, novel drug-eluting CS–LP nanocomposite coatings for potential orthopaedic applications were successfully prepared on the surface of titanium foils by EPD. The cytotoxicity of the composite films was evaluated by examining MG-63 osteoblast-like cells. Formation of intercalated clay particles embedded in the CS hydrogel was confirmed by X-ray and FTIR analysis. Thermogravimetric analysis indicated an enhanced thermal stability of the composite coatings as a result of the interaction of the polymer chains with the clay particles and formation of intercalated nanocomposite through penetration of CS chains into the galleries of the LP. In-vitro drug-release studies in PBS showed an initial burst release followed by a diffusion-controlled release up to 4 weeks. The controlled release of the antibiotic drug was attained by the intercalation of polymer and the drug macromolecules into silicate galleries. The clay particles were found to control the drug release rate of the coatings in PBS. The cellular results indicated that the presence of LP could be effective on the cell viability, attachment and ALP activity of MG-63 osteoblast-like cells. Analysis of cell proliferation by ALP expression showed enhancement of cell ALP enzyme activity because of the gradual dissolution LP into the culture medium as well as their effect on the adsorption process of biomolecules.
References
Shchipunov Y, Ivanova N, Silant’ev V. Bionanocomposites formed by in situ charged chitosan with clay. Green Chem. 2009;11:1758–61.
Kevadiya BD, Joshi GV, Mody HM, Bajaj HC. Biopolymer–clay hydrogel composites as drug carrier: host–guest intercalation and in vitro release study of lidocaine hydrochloride. Appl Clay Sci. 2011;52:364–7.
Ojijo V, Ray SS. Processing strategies in bionanocomposites. Prog Polym Sci. 2013;38:1543–89.
Pongjanyakul T, Khunawattanakul W, Strachan CJ, Gordond KC, Puttipipatkhachorn S, Rades T. Characterization of chitosan–magnesium aluminum silicate nanocomposite films for buccal delivery of nicotine. Int J Biol Macromol. 2013;55:24–31.
Croisier F, Jerome C. Chitosan-based biomaterials for tissue engineering. Eur Polym J. 2013;49(4):780–92.
Sarasam A, Brown P, Khajotia S, Dmytryk J, Madihally S. Antibacterial activity of chitosan-based matrices on oral pathogens. J Mater Sci. 2008;19(3):1083–90. doi:10.1007/s10856-007-3072-z.
Xie W, Xu P, Wang W, Liu Q. Preparation and antibacterial activity of a water-soluble chitosan derivative. Carbohydr Polym. 2002;50(1):35–40.
Pattnaik S, Nethala S, Tripathi A, Saravanan S, Moorthi A, Selvamurugan N. Chitosan scaffolds containing silicon dioxide and zirconia nano particles for bone tissue engineering. Int J Biol Macromol. 2011;49(5):1167–72.
Cai Q, Gu Z, Fu T, Liu Y, Song H, Li F. Kinetic study of chitosan degradation by an electrochemical process. Polym Bull. 2011;67(4):571–82.
Sinha VR, Singla AK, Wadhawan S, Kaushik R, Kumria R, Bansal K, et al. Chitosan microspheres as a potential carrier for drugs. Int J Pharm. 2004;274(1–2):1–33.
Di Martino A, Sittinger M, Risbud MV. Chitosan: a versatile biopolymer for orthopaedic tissue-engineering. Biomaterials. 2005;26(30):5983–90.
Bernkop-Schnürch A, Dünnhaupt S. Chitosan-based drug delivery systems. Eur J Pharm Biopharm. 2012;81:463–9.
Peter M, Binulal NS, Soumya S, Nair SV, Furuike T, Tamura H, et al. Nanocomposite scaffolds of bioactive glass ceramic nanoparticles disseminated chitosan matrix for tissue engineering applications. Carbohydr Polym. 2010;79(2):284–9.
Khunawattanakul W, Puttipipatkhachorn S, Rades T, Pongjanyakul T. Chitosan–magnesium aluminum silicate nanocomposite films: physicochemical characterization and drug permeability. Int J Pharm. 2010;393:219–29.
Cojocariu A, Profire L, Aflori M, Vasile C. In vitro drug release from chitosan/Cloisite 15A hydrogels. Appl Clay Sci. 2012;57:1–9.
Salcedo I, Aguzzi C, Sandri G, Bonferoni MC, Mori M, Cerezo P, et al. In vitro biocompatibility and mucoadhesion of montmorillonite chitosan nanocomposite: a new drug delivery. Appl Clay Sci. 2012;55:131–7.
Jung H, Kim H-M, Choy YB, Hwang S-J, Choy J-H. Laponite-based nanohybrid for enhanced solubility and controlled release of itraconazole. Int J Pharm. 2008;349:28–290.
Wang Q, Zhang J, Wang A. Alkali activation of halloysite for adsorption and release of ofloxacin. Appl Surf Sci. 2013;287:54–61.
Fan Q, Shan D, Xue H, He Y, Cosnier S. Amperometric phenol biosensor based on laponite clay–chitosan nanocomposite matrix. Biosens Bioelectron. 2007;22:816–21.
Gaharwar AK, Rivera CP, Wu C-J, Schmidt G. Transparent, elastomeric and tough hydrogels from poly(ethylene glycol) and silicate nanoparticles. Acta Biomater. 2011;7:4139–48.
Ghadiri M, Chrzanowski W, Lee WH, Fathi A, Dehghani F, Rohanizadeh R. Physico-chemical, mechanical and cytotoxicity characterizations of Laponite®/alginate nanocomposite. Appl Clay Sci. 2013;58:64–73.
Gaharwar AK, Schexnailder PJ, Kline BP, Schmidt G. Assessment of using Laponite® cross-linked poly(ethylene oxide) for controlled cell adhesion and mineralization. Acta Biomater. 2011;7:568–77.
Shi Q, Li Q, Shan D, Fan Q, Xue H. Biopolymer-clay nanoparticles composite system (Chitosan-laponite) for electrochemical sensing based on glucose oxidase. Mater Sci Eng C. 2008;28:1372–5.
Yang H, Hua S, Wang W, Wang A. Composite hydrogel beads based on chitosan and laponite: preparation, swelling, and drug release behaviour. Iran Polym J. 2011;20(6):479–90.
Esposito S, Leone S. Prosthetic joint infections: microbiology, diagnosis, management and prevention. Int J Antimicrob Agents. 2008;32(4):287–93.
Xie Z, Liu X, Jia W, Zhang C, Huang W, Wang J. Treatment of osteomyelitis and repair of bone defect by degradable bioactive borate glass releasing vancomycin. J Control. Release. 2009;139(2):118–26.
Goodman SB, Yao Z, Keeney M, Yang F. The future of biologic coatings for orthopaedic implants. Biomaterials. 2013;34(13):3174–83.
Corni I, Ryan MP, Boccaccini AR. Electrophoretic deposition: from traditional ceramics to nanotechnology. J Eur Ceram Soc. 2008;28(7):1353–67.
Boccaccini AR, Keim S, Ma R, Li Y, Zhitomirsky I. Electrophoretic deposition of biomaterials. J R Soc Interface. 2010;7(Suppl 5):S581–613.
Gebhardt F, Seuss S, Turhan MC, Hornberger H, Virtanen S, Boccaccini AR. Characterization of electrophoretic chitosan coatings on stainless steel. Mater Lett. 2012;66(1):302–4.
Simchi A, Pishbin F, Boccaccini AR. Electrophoretic deposition of chitosan. Mater Lett. 2009;63(26):2253–6.
Pishbin F, Simchi A, Ryan MP, Boccaccini AR. Electrophoretic deposition of chitosan/45S5 Bioglass® composite coatings for orthopaedic applications. Surf Coat Technol. 2011;205:5260–8.
Ordikhani F, Tamjid E, Simchi A. Characterization and antibacterial performance of electrodeposited chitosan-vancomycin composite coatings for prevention of implant-associated infections. Mater Sci Eng C. 2014;41:240–8.
Ordikhani F, Simchi A. Long-term antibiotic delivery by chitosan-based composite coatings with bone regenerative potential. Appl Surf Sci. 2014;317:56–66.
Ordikhani F, Farani MR, Dehghani M, Tamjid E, Simchi A. Physicochemical and biological properties of electrodeposited graphene oxide/chitosan films with drug-eluting capacity. Carbon. 2015;84:91–102.
Chakraborty SP, Pramanik P, Roy S. A review on emergence of antibioticresistant staphylococcus aureus and role of chitosan nanoparicle in drug delivery. Int J Life Sci Pharma Res. 2012;2:L96–115.
Chakraborty SP, Sahu SK, Pramanik P, Roy S. In vitro antimicrobial activity of nanoconjugated vancomycin against drug resistant Staphylococcus aureus. Int J Pharm. 2012;436(1–2):659–76.
Xu Y, Ren X, Hanna MA. Chitosan/clay nanocomposite film preparation and characterization. J Appl Polym Sci. 2006;99:1684–91.
Besra L, Liu M. A review on fundamentals and applications of electrophoretic deposition (EPD). Prog Mater Sci. 2007;52(1):1–61.
Yuan Q, Shah J, Hein S, Misra RDK. Controlled and extended drug release behavior of chitosan-based nanoparticle carrier. Acta Biomater. 2010;6(3):1140–8.
Ponsonnet L, Reybier K, Jaffrezic N, Comte V, Lagneau C, Lissac M, et al. Relationship between surface properties (roughness, wettability) of titanium and titanium alloys and cell behaviour. Mater Sci Eng C. 2003;23(4):551–60.
Vlacic-Zischke J, Hamlet SM, Friis T, Tonetti MS, Ivanovski S. The influence of surface microroughness and hydrophilicity of titanium on the up-regulation of TGFβ/BMP signalling in osteoblasts. Biomaterials. 2011;32(3):665–71.
Pishbin F, Mourino V, Gilchrist JB, McComb DW, Kreppel S, Salih V, et al. Single-step electrochemical deposition of antimicrobial orthopaedic coatings based on a bioactive glass/chitosan/nano-silver composite system. Acta Biomater. 2013;9(7):7469–79.
Depan D, Kumar AP, Singh RP. Cell proliferation and controlled drug release studies of nanohybrids based on chitosan-g-lactic acid and montmorillonite. Acta Biomater. 2009;5:93–100.
Gaharwar AK, Schexnailder PJ, Jin Q, Wu C-J, Schmidt G. Addition of chitosan to silicate cross-linked PEO for tuning osteoblast cell adhesion and mineralization. Appl Mater Interfaces. 2010;2:3119–27.
Pon-On W, Charoenphandhu N, Teerapornpuntakit J, Thongbunchoo J, Krishnamra N, Tang IM. In vitro study of vancomycin release and osteoblast-like cell growth on structured calcium phosphate-collagen. Mater Sci Eng C. 2013;33(3):1423–31.
Cheng Y, Luo X, Bentley WE, Betz J, Rubloff GW, Buckhout-White S, et al. In situ quantitative visualization and characterization of chitosan electrodeposition with paired sidewall electrodes. Soft Matter. 2010;6:3177–83.
Ghadiri M, Hau H, Chrzanowski W, Agus H, Rohanizadeh R. Laponite clay as a carrier for in situ delivery of tetracycline. RSC Adv. 2013;3:20193–201.
Davies JE, Ajami E, Moineddin R, Mendes VC. The roles of different scale ranges of surface implant topography on the stability of the bone/implant interface. Biomaterials. 2013;34(14):3535–46.
Mieszawska AJ, Fourligas N, Georgakoudi I, Ouhib NM, Belton DJ, Perry CC, et al. Osteoinductive silk-silica composite biomaterials for bone regeneration. Biomaterials. 2010;31(34):8902–10.
Acknowledgments
The authors thank funding support from the Grant Program of Sharif University of Technology (No. G930305) and Elite National Institute (No. ENL 5418).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ordikhani, F., Dehghani, M. & Simchi, A. Antibiotic-loaded chitosan–Laponite films for local drug delivery by titanium implants: cell proliferation and drug release studies. J Mater Sci: Mater Med 26, 269 (2015). https://doi.org/10.1007/s10856-015-5606-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-015-5606-0