
Abstract
Nanocrystalline calcium phosphate apatites are biomimetic compounds analogous to bone mineral and are at the origin of the bioactivity of most biomaterials used as bone substitutes. Their unique surface reactivity originates from the presence of a hydrated layer containing labile ions (mostly divalent ones). So the setup of 3D biocompatible apatite-based bioceramics exhibiting a high reactivity requests the development of «low» temperature consolidation processes such as spark plasma sintering (SPS), in order to preserve the characteristics of the hydrated nanocrystals. However, mechanical performances may still need to be improved for such nanocrystalline apatite bioceramics, especially in view of load-bearing applications. The reinforcement by association with biopolymers represents an appealing approach, while preserving the advantageous biological properties of biomimetic apatites. Herein, we report the preparation of composites based on biomimetic apatite associated with various quantities of microcrystalline cellulose (MCC, 1–20 wt%), a natural fibrous polymer. The SPS-consolidated composites were analyzed from both physicochemical (X-ray diffraction, Fourier transform infrared, solid state NMR) and mechanical (Brazilian test) viewpoints. The preservation of the physicochemical characteristics of apatite and cellulose in the final material was observed. Mechanical properties of the composite materials were found to be directly related to the polymer/apatite ratios and a maximum crushing strength was reached for 10 wt% of MCC.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In the field of orthopedic or dental materials, bioceramics have been used for the last decades, exploiting their chemical inertia or bioactivity, and their osteoconduction properties [1], despite their moderate mechanical resistance.
Among bioceramics, those based on calcium phosphates and more specifically (hydroxy)apatite [2] are the most commonly encountered due to their compositional and structural similarities with the mineral part of calcified tissues. In recent years, many studies have been conducted more specifically on biomimetic nanocrystalline apatites that exhibit physicochemical characteristics very close to those of bone mineral; they are characterized by constitutive crystals in the nanometer range, as well as a metastable and hydrated feature and have a non-stoichiometric composition [3, 4]. However, despite these characteristics, hydroxyapatite and its nanocrystalline counterparts (more reactive and less stable) are not considered as effective solutions for the realization of bulk materials for orthopedic/dental uses in load-bearing sites, due to difficulties of material shaping and limited mechanical properties [1].
The methods developed for bioceramics shaping generally involve high temperatures, resulting in significant grain coarsening and reduction in surface reactivity/bioactivity. Therefore, the use of fast or “flash” processing methods at moderate sintering temperatures, such as spark plasma sintering (SPS), has been investigated and proved to be especially appealing [5, 6]. This technique has been shown to preserve most of the characteristics of thermally unstable biomimetic nanocrystalline apatites in sintered bodies. Indeed, according to the literature [5, 6], biomimetic apatite 3D scaffolds obtained by this technique are characterized by very encouraging biological properties. However, they also show limited mechanical features. One possible solution to solve this type of problem would be the association of organic and fibrous materials, such as biocompatible polymers, to the ceramic matrix. In the case of calcium phosphates for bone substitution, such reinforcements with polymers are mainly encountered in bone filling applications like in the formulation of injectable cements [7]. However, it appears difficult to use organic materials for the development of bulk ceramics processed by conventional sintering methods using high temperatures (e.g., 1000 °C), because the degradation of polymers at these temperatures may lead to the loss of the expected functional properties of the composite. The consolidation of biomimetic apatite at “low” temperature by SPS may allow the use of a wide variety of (biocompatible) polymers which require “low” temperature processing due to their domain of thermal stability.
Among such polymers, cellulose is an appealing candidate. It is a linear homopolymer of glucose, which is the polymeric material most widely encountered/distributed in nature. Furthermore, the established biocompatibility of cellulose and its derivatives has promoted their use in many biomedical applications. Among the various cellulosic compounds employed, we selected microcrystalline cellulose (MCC) for its interesting characteristics [8–10]: (i) an excellent thermal stability (>200 °C), (ii) a fibrous/filamentous organization of the cellulose microcrystals, (iii) an elongated and irregular particle shape and (iv) high specific strength and modulus. All these features make it a prime candidate for the reinforcement of composite organic–inorganic materials, and it was thus chosen in this study.
2 Materials and methods
2.1 Powder synthesis
2.1.1 Biomimetic apatite
Non-carbonated nanocrystalline apatites with Ca/P close to 1.5, Ca10−x (PO4)6−x (HPO4) x (OH)2−x , were synthesized by double decomposition [3, 4] from a calcium nitrate solution (Ca(NO3)2·4H2O) and a solution of di-ammonium hydrogen phosphate (NH4)2HPO4 in excess. The calcium solution was quickly poured into the phosphate solution at room temperature (20 °C) before being stirred for a few minutes. The large excess of phosphate ions in the solution (relative to the Ca/P stoichiometry of hydroxyapatite) allows maintaining the reaction medium close to the physiological pH of 7.4 during the entire synthesis thanks to phosphate buffering. After the initial precipitation, the apatite samples were matured in the stock solution (aging) for 1 day at room temperature, without stirring (close to biomimetic conditions). During the maturation stage, the vials were hermetically closed to limit the incorporation of atmospheric CO2. After maturation [3], the precipitate was filtered, washed with deionized water, freeze-dried, and stored at low temperature (−18 °C); these last two steps being aimed at limiting any possible alterations of the compounds prior to analysis.
2.1.2 Microcrystalline cellulose
MCC is available in different pharmaceutical grades varying mainly by their bulk density, particle size, and moisture content. The MCC selected for this study is the VIVAPUR®102 (JRS Pharma). This material is characterized by an average particle size of 90–150 μ, a maximum moisture content of 7 % and a density of 0.28–0.33 g cm−3.
2.1.3 Composite materials
Different apatite and MCC blends were prepared with a three-dimensional Turbula T2C mixer (WAB, Switzerland) for 10 min at room temperature. The apatite-polymer mixtures produced corresponded to 0, 2, 10 and 20 % by weight of MCC, which will be respectively referred to in the text as Mix-0 %, Mix-2 %, Mix-10 % and Mix-20 %.
2.2 Powder processing via SPS consolidation (spark plasma sintering)
Powder consolidation was carried out in this work by SPS using an SPS 2080 Sumitomo coal mining equipment of the Plateforme Nationale CNRS de Frittage Flash (PNF2/CNRS) located at the University Toulouse III Paul Sabatier. This device is composed of a uniaxial press (up to 200 kN) and a maximal power supply of 8000 A and 10 V of DC-pulsed current. In our experiments, we limited the current to 4000 A, and the sequencing of pulses was 12:2 (12 pulses of 3.3 ms followed by two dead times without current of 3.3 ms) according to previous studies on SPS of biomimetic apatite [5]. The powder (0.63 g) was placed in a graphite mold of 15 mm diameter. To facilitate the disassembling, graphite sheets (Papyex®) were used. The mold containing the powder was introduced into the chamber of the device and a low mechanical load was applied to obtain a satisfactory electrical contact. The green density in the case of the pure apatite powder was close to 0.35 g cm−3 (relative density ~13 %). The processing chamber was then purged twice under vacuum and then a pressure of 1 atm of argon was maintained throughout the process. The temperature and pressure programs used in the sequence [5] are described in Fig. 1. The temperature was measured using a K type thermocouple located at 3 mm inside the die from the lateral surface, at equal distances from the top and bottom parts of the die. The full die geometry was as follows: inner diameter 15 mm, external diameter 40 mm and height 30 mm. The temperature distribution inside the sample cannot be easily predicted due to the thermal insulating characteristics of apatite materials and the chemical reactions (essentially endothermic) occurring during the sintering process. Faint temperature fluctuations (1–2 °C) around the set temperature were observed during dwelling time for the first 0.5 min, in line with our previous work on SPS sintering of biomimetic apatite [5].

Schematic, temperature and pressure programmed sequences for SPS
2.3 Physicochemical and microstructural characterization
The powders and samples obtained in this work were characterized by complementary techniques.
X-ray diffraction (XRD) was performed with a SIEMENS D5000 diffractometer using the CuKα1Kα2 radiation.
Vibrational spectrometry analysis was carried out using a Nicolet 5700 Fourier transform infrared (FTIR) spectrometer. Spectra were recorded in the 400–4000 cm−1 range, with a resolution of 4 cm−1, and using the KBr pellet method.
The microstructure of the consolidated samples was observed using a LEO 435 VP scanning electron microscope on sections of solid samples diametrically fractured and without any special sample preparation (no preliminary metal coating procedure). SEM micrographs were obtained by detecting either secondary electrons (SE) or backscattered electrons (BSE), the latter providing chemical contrast. The phase containing the heaviest elements (biomimetic apatite in the present case) is expected to appear clearer on the micrographs. Cellulose, containing lighter elements, is on the contrary expected to appear darker.
Specific surface areas were measured by the Brunauer–Emmett–Teller (BET) method with a Quantachrome Monosorb apparatus using nitrogen gaz.
2.4 Solid state NMR experiments
31P magic angle spinning (MAS) solid state NMR spectra were recorded at 14.1 T on a Varian VNMRS 600 MHz NMR spectrometer, using a 3.2 mm Varian T3 HXY MAS probe spinning at 20 kHz. 31P MAS NMR spectra of nanocrystalline apatite and SPS-20 % (the explanation on this sample notation is provided later in the article) were recorded using a 3 μs 90° excitation pulse (preceded by a train of presaturation pulses), and 100 kHz spinal-64 1H decoupling during acquisition. Four transients were acquired with a recycle delay of 64 s. Although this does not correspond to a full relaxation of the 31P resonances, no change in line shape was observed at lower recycle delays (e.g., 32 s). 1H → 31P cross-polarization (CP) MAS NMR experiments were also carried out to probe the spatial proximities between 1H and 31P nuclei. 1D CPMAS spectra were recorded using contact times (CT) of 0.5, 2 and 6 ms. Eight to 40 transients were acquired, with a recycle delays of 10 s. All 31P NMR spectra were referenced to synthetic hexagonal hydroxyapatite model compound (phosphate resonance at 2.8 ppm with respect to concentrated H3PO4).
Two-dimensional 1H → 31P CPMAS spectra were recorded 9.4 T on a Varian VNMRS 400 MHz spectrometer, using a 3.2 mm Varian T3 HXY MAS probe spinning at 20 kHz. Two different experiments were performed for each sample, using CT of 0.5 and 5 ms. The States acquisition method was used, and 12 acquisitions were co-added for each of the t 1 slices. The F 1 and F 2 spectral widths were 80 and 40 kHz, respectively. The recycle delays were set to 10 s. On the 2D spectra, the cross peaks which correlate 1H and 31P resonances help identify which 1H and 31P atoms are spatially close to each other in the material.
1H MAS NMR spectra were recorded at 14.1 T on a Varian VNMRS 600 MHz NMR spectrometer, using a 3.2 mm Varian T3 HXY MAS probe spinning at 20 kHz. A spin-echo sequence was used, with a half-echo delay corresponding to four rotor periods (τ ~ 200 μs). A total of eight transients were acquired, with a recycle delay of 10 s (ensuring full relaxation). All 1H NMR spectra were referenced to adamantane (1.8 ppm with respect to tetramethylsilane—TMS).
1H → 13C CP MAS NMR experiments were carried out at 14.1 T on a varian VNMRS 600 MHz spectrometer, using a 3.2 mm varian T3 HXY MAS probe spinning at 10 kHz. A CT of 4 ms was used, and spinal 64 1H decoupling (100 kHz RF) was applied during acquisition. The recycle delay was set to 4 s. A total of 800 and 10,000 transients were acquired for the MCC and SPS-20 % samples, respectively. All 13C NMR spectra were referenced to adamantane (high frequency signal at 38.5 ppm with respect to TMS).
A 13C{31P} REDOR (rotational echo double resonance) NMR pulse sequence was used to try to characterize the organic-mineral interface in one of the cellulose/apatite composites. The experiment was performed at 14.1 T on a varian VNMRS 600 MHz NMR spectrometer, using a 3.2 mm varian T3 HXY MAS probe spinning at 10 kHz. The same CP conditions as in the 1H–13C CPMAS experiments were used. Total dephasing times of 10, 20, and 30 ms were successively tested, using 8 μs rotor-synchronized π pulses on 31P. Spinal 64 1H decoupling (100 kHz RF) was used during the dephasing and acquisition periods. A total of ~3000 scans were acquired.
2.5 Mechanical characterization
Standard mechanical measurements for orthopedic applications require cylinder compressive testing with specific ratio between length and diameters (length ≥ 3 radius). This ratio cannot be produced easily because the green density (≈0.3 g cm−3) is very low compare to the final density (≈1.8 g cm−3) and we cannot fill the standard die more than what was performed here. For this reason, the mechanical resistance of the consolidated scaffolds was tested by way of the Brazilian test, aka splitting tension test, allowing evaluating the tensile strength σt of the ceramics. In this mechanical testing method, the cylinder (or pellet) is compressed vertically, across one diameter, between the plates of a press. This test is valid only if the resulting fracture is caused by a tensile stress, rather than by a compressive force or shear. The force causing failure can be identified visually by a central straight-through crack along the loading direction. In a successful test, the fracture plane therefore divides the cylinder into two halves.
3 Results and discussion
3.1 Raw materials
3.1.1 Biomimetic apatite
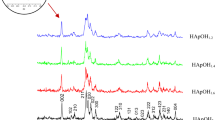
The purity of the synthesized nanocrystalline apatite powder was assessed by XRD (Fig. 2b). The diffraction pattern obtained was found to be similar to those obtained in previous work [5], and corresponds to a nanocrystalline apatite phase, characterized by a low degree of crystallinity as in the mineral phase of bone. The diffraction peaks can be indexed in the hexagonal apatitic system (space group P63/m) in accordance with the ICCD PDF card no. 9-432 relative to hydroxyapatite. The broadness of the peaks points to the low crystallinity of the samples which is related both to the nanometer-scale dimensions of the crystallites and to crystal disorder as was investigated previously [11]. The existence of preferential crystallite orientations (i.e., texture), which highlights the dimensional anisotropy of the grains, can also be pointed out, for example, with peaks (002) and (004) or (00 l) being thinner than peaks (hk0). Nanometric crystal dimensions and anisotropy of biomimetic apatite were also observed in other works on the basis of electron microscopy analyses [5]. The specific surface area measured by BET is relatively high and close to 150 m2 g−1.

Diffraction patterns of (a) MCC powder, (b) biomimetic apatite powder, (c) sample MIX-2 %, (d) sample SPS-2 %. Indexed peaks correspond to hydroxyapatite and those with asterisk to MCC
The Ca/P molar ratio determined from XRD data after calcination at 1000 °C for 15 h [3] was found to be close to 1.51. This value is significantly lower than 1.67, which is the value observed in stoichiometric hydroxyapatite. This result confirms the non-stoichiometric character of the apatitic phase prepared in this work.
The FTIR spectrum recorded on the starting powder of apatite (Fig. 3b) shows the characteristic bands of nanocrystalline non-stoichiometric apatite (non-carbonated) [3, 4] with (i) characteristic phosphate bands at 474 cm−1 (ν2 PO4 3−), 570 and 601 cm−1 (ν4 PO4 3−), 960 cm−1 (ν1 PO4 3−), 1030 and 1081 cm−1 (ν3 PO4 3−), 1140–1150, 1250 and 875 cm−1 (ν HPO4 2−) (ii) the difficult detection of absorption bands related to apatitic OH− ions, observed in biomimetic apatite at 632 and 3572 cm−1 and (iii) the observation of water bands around 1640 cm−1 and between 3000 and 3400 cm−1 (broad band). Special attention was paid to the analysis of the absorption domain around the vibration mode ν4 PO4 3− (from 570 to 601 cm−1). Indeed, in this region, previous studies have shown that not only the bands typical of apatitic phosphate or OH vibrations occurred, but also additional bands related to non-apatitic surface phosphate ions [3, 11]. These non-apatitic chemical environments have been linked to the presence of a structured hydrated (and ionic) layer at the surface of the nanocrystals [4]. This layer plays a key role in the biological activity of such apatites (for both natural biological apatites and their biomimetic synthetic counterparts): it is for example involved in surface ion exchanges. Its conservation and systematic characterization will thus be necessary at each step of the process described below, to verify that the “hydrated layer” has not been destructed.

FTIR spectra of (a) MCC powder, (b) biomimetic apatite powder, (c) sample MIX-2 %, (d) sample SPS-2 %. (at ×30, ×200 or ×2K magnifications)
Solid state NMR was also used to characterize the hydroxyapatite precursor (Fig. 4). The 31P NMR spectrum shows one main signal centered at ~2.9 ppm, and a broad underlying component at lower frequencies. The presence of different phosphate contributions is not surprising considering the IR spectrum of this phase. Based on previous 31P NMR characterizations of hydroxyapatite (HA) [12, 13], the narrow signal at 2.9 ppm can be assigned to phosphates in the bulk of the HA crystal lattice. The other phosphate resonance corresponds to hydrogen phosphate anions, as shown by additional 1D and 2D 1H → 31P CPMAS experiments (Figs. 5, 6). Indeed, in the 1D CPMAS spectra recorded using different CT, the relative intensity of the broad component is predominant at the shortest CT (CT = 500 μs). Moreover, in the 2D CPMAS NMR spectrum (Fig. 6), it appears that this broad phosphate peak mainly correlates with 1H resonances ranging from ~3 to 17 ppm, which can be assigned to protons of HPO4 2− anions and water molecules. Based on these characterizations and on previous studies on nanocrystalline apatites prepared using the exact same synthetic procedure [5, 14], it can be suggested that the broad low-frequency 31P resonance mainly corresponds to the non-apatitic hydrogen phosphates present in the hydrated layer at the surface of the apatite nanocrystals. It is worth noting that similar 2D CPMAS spectra have been reported in the literature for nanocrystalline apatite-chondroitin sulfate composites [15].

a Single-pulse 31P MAS NMR spectra of biomimetic apatite and SPS-20 %. b Hahn-echo 1H-NMR spectra of biomimetic apatite, MCC and SPS-20 %. c 13C CPMAS NMR spectra of MCC and SPS-20 %, with the assignment of the different C resonances (the diamond signals correspond to an amorphous component—see Ref. [17]). For all spectra, measurement conditions can be found in the experimental section of the manuscript

31P MAS NMR spectra (in black) and CPMAS NMR spectra (in grey) of the a biomimetic apatite and b SPS-20 % samples, recorded at 14.1 T

1H–31P 2D CPMAS NMR experiments, recorded at 9.4 T, using a contact time of 500 μs. The projections shown here correspond to the sum of the individual slices
3.1.2 Microcrystalline cellulose
The treatment for obtaining MCC produces agglomerates having a higher degree of crystallinity than the original cellulose. The XRD pattern obtained for the MCC used in this work is consistent with what may be found in literature (Fig. 2a). The main single peak around 22.6° is characteristic of crystalline cellulose I [8, 10]. The FTIR spectrum of the commercial MCC powder presents bands usually attributed to this cellulose (Fig. 3a), e.g., at 2900 cm−1 for symmetric stretching vibrations of C–H, ~1060 cm−1 due to C–O/C–C stretching [10, 16] or 3450 cm−1 corresponding to O–H stretching of hydroxyl groups.
The 13C CPMAS NMR spectrum of MCC presents the signals characteristic of a cellulose phase [17, 18]. More specifically, the position of the C6 peak at ~65.7 ppm demonstrates the presence of type-I cellulose in the sample, in line with the XRD data [17, 19]. Amorphous components are also detected, as often observed on NMR spectra of nanocrystalline cellulose [20].
3.1.3 Powder mixing
In the mixed powder, no damage of the precursors was observed (based on XRD and IR analyses), thanks to the gentle mixing conditions used with the Turbula. It should however be noted that the XRD characterization of the MCC phase in these mixtures was delicate, in particular due to the semi-crystallinity of MCC. The similar size (around 100 μ) of the agglomerates in both powders (apatite and MCC) favored the production of a relatively homogeneous mixture, which was then maintained during the SPS consolidation. This homogeneous powder mixture is clearly visible on the sectional micrographs of the pellets (Fig. 7).

SEM micrographs (cross section) performed on samples SPS-2 % and SPS-20 % at various magnifications
3.2 SPS consolidation
3.2.1 Physicochemical characterizations
The influence of the weight percentage of cellulose associated to the apatite matrix was investigated in this paper with the following compositions: 0, 2, 10, 20 %. Between 11 and 16 samples for each composition were prepared, and they are referred to as SPS-“wt% cellulose”.
After SPS processing, the analysis by XRD suggested, by the slight modification of the signal around 22°, the presence of both the apatite and MCC phases. The IR spectra obtained on SPS samples were also found to be very similar to the spectra of the mixed powders (Fig. 3). A detailed analysis of the ν4 PO4 vibration domain was done as explained previously [11], indicating in particular the presence of a characteristic shoulder between 500 and 550 cm−1 which is attributed to non-apatitic HPO4 2− ions, confirming the persistence of the hydrated layer on the apatite nanocrystals contained in our samples after SPS. A small bump at about 632 cm−1 assigned to OH− ions in hydroxylated apatites was also evidenced after SPS treatment. These observations thus reveal a slight change in the composition of the apatite phase, which may be explained by some degree of internal hydrolysis of phosphate ions, due to the SPS processing, according to a reaction of the type:
H2O + PO4 3− → OH− + HPO4 2−.
It is worth noting that for both the powder mixture and SPS-treated samples, it remains difficult to observe the organic part in the composite material using FTIR or XRD analyses, even at high concentrations of MCC.
The SPS-consolidated samples with the highest cellulose content were then analyzed by solid state NMR for complementary characterizations, especially to provide unambiguous evidence of the integrity of the cellulose component. The purpose of the NMR analyses was also to probe the structure of the material at the organic-mineral interface using 13C···31P correlation experiments. The 1H, 13C and 31P solid state NMR spectra of the SPS-20 % sample are shown in Figs. 4, 5 and 6.
As may be seen in Fig. 4, the 13C CPMAS NMR spectra of SPS-20 % and MCC are very similar, bringing direct evidence that the bulk structure of the cellulose crystallites was not affected by the SPS treatment. Both spectra depict the signals characteristic of a cellulose phase, with the same relative intensity of signals for the amorphous and crystalline domains [17, 18]. Moreover, the position of the C6 peak at 65.7 ppm demonstrates that type-I cellulose is preserved after SPS treatment [17, 19].
The single-pulse 31P MAS NMR spectra of the SPS-20 % sample and of the biomimetic apatite phase used for its synthesis are also very similar (Figs. 4, 5, 6). The signal characteristic of the non-apatitic environments is still present, which confirms that the conditions used for the SPS treatment did not destroy the hydrated layer. Moreover, a careful comparison of the spectra recorded by direct excitation (Fig. 4) shows that the relative intensity of this signal has even slightly increased after SPS. This demonstrates that other small changes in the apatite component can occur upon SPS treatment, in addition to those already evidenced by IR on the SPS-2 % sample.
The 1D CPMAS spectra recorded at 500 μs for SPS-20 % and biomimetic apatite samples were then fitted using two components, one centered at 2.9 ppm, and the other at lower frequencies (see Fig. S1 in Online Ressource). After fitting, it appears that the position and full-width at half maximum of the broad peak have changed, with an increase in full-width at half maximum in the SPS-20 % sample. This could be seen as a direct consequence of the interaction of the hydrated layer with the surface of the cellulose crystallites, which would lead to a larger distribution of 31P local environments for the non-apatitic phosphates. An attempt was then made to determine which carbon atoms of the cellulose polymer are closest to the mineral surface, using the 13C{31P} REDOR sequence [21, 22]. However, even for total recoupling times of 30 ms, no dephasing of the 13C resonances was observed (data not shown). Several explanations can be proposed for the absence of dephasing, such as: (i) the large size of the cellulose crystallites (~100 μm in average), the 13C NMR signals observed mainly arise from the core of the crystallites, and those coming from the carbon atoms close to the surface only represent a very small fraction of the overall 13C intensity, meaning that small dephasings of the surface 13C resonances would be very difficult to observe; (ii) the change in transverse relaxation of the 13C atoms at the interface, which hampers the detection of 13C–31P proximities using REDOR dephasing times such as those used here (which correspond to the dephasing times classically used for studying the organic-mineral interface in bone) [21–24].
3.2.2 Micro- and macro-structural characterizations
The apparent density of the consolidated samples, depending on the composition, is shown in Fig. 8. On the graph of the density versus composition, a small increase in density is visible, followed by a slow decrease for compositions beyond 2 % of MCC. The density was comprised between 1.8 and 1.9 g cm−3, corresponding to about 75 % densification. The consolidated samples of apatite powders mixed with cellulose (wt%) were observed by SEM at different magnifications (Fig. 7). At low magnification (×30), a homogeneous and dense matrix was observed. Lens-shaped cellulose aggregates (observable in dark in the backscattered electron mode, BSE) with an approximate length of 100 μm, are found to be homogeneously dispersed and oriented parallel to the surface of the pellets. The surface fraction of the cellulose aggregates increases with the wt% of cellulose while maintaining a homogeneous spatial distribution. By increasing the magnification (×2000), the analysis of such lens-shaped cellulose aggregates indicates that the contact zone between the MCC and apatite presents no apparent cracks. According to all data provided by the observations, we can deduce that the cellulose is distributed in the mineral matrix homogeneously and is in “intimate” contact with the latter.

Tensile stress and density versus wt % cellulose. Insert: macropictures of samples SPS-10 %, SPS-20 % after mechanical characterizations
The XRD analysis (Fig. 2) on the surface of the consolidated samples gives us information about the microstructure. The broadness of XRD peaks reveals the low crystallinity and/or nanosize of the particles after SPS consolidation, as for the starting powder mixtures. The peak widths are also dependent on the crystallographic orientation, which emphasizes the anisotropy of grain size [peaks (002) (004) or (00l) are narrower than the (hk0)]. The measured peak intensities differ from those given by the JCPDS No. 9-432, in relation with non-stoichiometry and/or texturing after SPS consolidation. Preferred orientations appear detectable especially by examination of the 2θ peaks at 26° (002), 33° (300), 39.5° (310) and 53.5° (004). These XRD patterns suggest a preferential orientation corresponding to an alignment of the c-axis of the apatite lattice, associated with the elongation direction of the platelet crystals, parallel to the diametrical surface of the pellet. The mechanical pressure applied during uniaxial SPS processing, associated with an anisotropic grain size, can indeed promote such a texture (preferred orientation).
3.2.3 Mechanical evaluations
SPS-consolidated samples corresponding to each tested composition were characterized mechanically using the Brazilian test previously described (ten pellets tested for each mixing ratio). A visual inspection of fracture allowed validating the test for each pellet (see Fig. 8 insert).
No variation of the maximum crushing stress was found between the first two compositions (0 and 2 wt% of cellulose). In contrast, it almost doubled for a proportion of 10 wt% of MCC while maintaining approximately the same density. For a proportion of 20 wt% of MCC, a scaling phenomenon was observed rather than a fracture phenomenon, rendering invalid an evaluation via Brazilian test.
These findings indicate that, generally speaking, the apatite matrix clearly gets reinforced mechanically thanks to the association with MCC. However, the change in mechanical behavior is seen to depend on the quantity of MCC in close interaction with apatite grains. Beyond a certain amount of MCC, no fracture in the apatitic matrix was evidenced, but a scaling phenomenon appeared. This type of “fragmentation” could thus be due to a too large number of MCC aggregates in the composites, creating mechanical weaknesses. This observation points to the existence of a limit in the amount of MCC to be associated to biomimetic apatite in view of conferring added flexibility while retaining the rigidity due to apatite.
4 Conclusion
For the first time, composite biomaterials based on polymerceramic matrix were consolidated at low temperature (150 °C). This technological leap has been made possible by the use of a highly reactive powder as component of the matrix, composed of biomimetic apatite, and a polymer of high thermal stability, MCC, and by their association using a flash consolidation process: SPS. Complementary physicochemical analyses before and after SPS processing confirmed the absence of significant degradation of the two starting components (biomimetic apatite and MCC), thus rendering these composite materials promising in view of applications as biomaterials exploiting the intrinsic biological properties of each initial component. Mechanical evaluations (by way of Brazilian tests) were carried out on consolidated samples obtained with increasing amounts of MCC. Pellets prepared with 10 wt% MCC exhibited a clearly improved mechanical behavior. However, the amount of MCC directly influenced the mechanical properties: beyond a certain limit of MCC—between 10 and 20 wt% in our case—the rigidity of the apatite matrix was progressively lost in favor of a delamination phenomenon. The analysis of the interface between apatite and cellulose by way of SEM allowed us however to point out in all cases a close interaction between the two components of the composites. The role of water, unveiled for example by FTIR, will have to be further inspected for a more detailed understanding of the consolidation of such composites by SPS.
References
Billotte WG. Ceramic biomaterials. In: Park JB, Bronzino JD, editors. Biomaterials principles and applications. Boca Raton: CRC Press; 2003. p. 21–54.
Heness G, Ben-Nissan B. Innovative bioceramics. Mater Forum. 2004;27:104–14.
Eichert D. Etude de la réactivité de surface d’apatites de synthèse nanocristallines. PhD thesis, INP Toulouse, 2001.
Cazalbou S, Eichert D, Ranz X, Drouet C, Combes C, Harmand MF, Rey C. Ion exchanges in apatites for biomedical application. J Mater Sci. 2005;16:405–9.
Grossin D, Rollin-Martinet S, Estournes C, Rossignol F, Champion E, Combes C, Rey C, Chevallier G, Drouet C. Biomimetic apatite sintered at very low temperature by spark plasma sintering: physico-chemistry and microstructure aspects. Acta Biomater. 2010;6:577–85.
Drouet C, Bosc F, Banu M, Largeot C, Combes C, Dechambre G, Estournes C, Raimbeaux G, Rey C. Nanocrystalline apatites: from powders to biomaterials. Powder Technol. 2009;190:118–22.
Mano JF, Sousa RA, Boesel LF, Neves NM, Reis RL. Bioinert, biodegradable and injectable polymeric matrix composites for hard tissue replacement: state of the art and recent developments. Compos Sci Technol. 2004;64:789–817.
Tobyn M, McCarthy GP, Staniforth JN, Edge S. Physicochemical comparison between microcrystalline cellulose and silicified microcrystalline cellulose. Int J Pharm. 1998;169:183–94.
Kiziltas A, Gardner DJ, Han Y, Yang H-S. Dynamic mechanical behavior and thermal properties of microcrystalline cellulose (MCC)-filled nylon 6 composites. Thermochim Acta. 2011;519:38–43.
Adel AM, El-Wahab ZHA, Ibrahim AA, Al-Shemy MT. Characterization of microcrystalline cellulose prepared from lignocellulosic materials. Part II: physicochemical properties. Carbohydr Polym. 2011;83:676–87.
Vandecandelaere N, Rey C, Drouet C. Biomimetic apatite-based biomaterials: on the critical impact of synthesis and post-synthesis parameters. J Mater Sci. 2012;23:2593–606.
Elliott C. Structure and chemistry of the apatites and other calcium orthophosphates, studies in inorganic chemistry, vol. 18. Amsterdam: Elsevier; 1994. p. 288.
Kolodziejski W. Solid-state NMR studies of bones. Top Curr Chem. 2004;246:235–70.
Eichert D, Drouet C, Sfihi H, Rey C, Combes C. Nanocrystalline apatite-based biomaterials: synthesis, processing and characterization. In: Kendall JB, editor. Biomaterials research advances. New York: Nova Science Publishers; 2007. p. 93–143.
Bradley JV, Bridgland LN, Colyer DE, Duer MJ, Friscic T, Gallagher JR, Reid DG, Skepper JN, Trasler CM. NMR of biopolymer-apatite composites: developing a model of the molecular structure of the mineral-matrix interface in calcium phosphate biomaterials. Chem Mater. 2010;22:6109–16.
Rojas J, Lopez A, Guisao S, Ortiz C. Evaluation of several microcrystalline celluloses obtained from agricultural by-products. J Adv Pharm Technol Res. 2011;2:144–50.
Atalla RH, Gast JC, Sindorf DW, Bartuska JC, Maciel GE. Carbon-13 NMR spectra of cellulose polymorphs. J Am Chem Soc. 1980;102:3249–51.
Park S, Johnson DK, Ishikawa CI, Parilla PA, Davis MF. Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose. 2009;16:641–7.
Isogai A, Usuda M, Kato T, Uryu T, Atalla RH. Solid-state CP/MAS carbon-13 NMR study of cellulose polymorphs. Macromolecules. 1989;22:3168–72.
Idstrom A, Brelid H, Nydén M, Nordstierna L. CP/MAS 13C NMR study of pulp hornification using nanocrystalline cellulose as a model system. Carbohydr Polym. 2013;92:881–4.
Jaeger C, Groom NS, Bowe EA, Horner A, Davies ME, Murray RC, Duer MJ. Investigation of the nature of the protein-mineral interface in bone by solid-state NMR. Chem Mater. 2005;17:3059–61.
Nikel O, Laurencin D, Bonhomme C, Sroga GE, Besdo S, Lorenz A, Vashishth D. Solid state NMR investigation of intact human bone quality: balancing issues and insight into the structure at the organic-mineral interface. J Phys Chem C. 2012;116:6320–31.
Nikel O, Laurencin D, McCallum SA, Gundberg CM, Vashishth D. NMR investigation of the role of osteocalcin and osteopontin at the organic–inorganic interface in bone. Langmuir. 2013;29:13873–82.
Bonhomme C, Gervais C, Laurencin D. Recent NMR developments applied to organic–inorganic materials. Prog Nucl Magn Reson Spectrosc. 2014;77:1–48.
Acknowledgments
This research was partly funded by the French National Research Agency (ANR) through the NanoBioCer Project (ANR-07-BLAN-0373). We are grateful to Makram EL-BACHAWATI for the preliminary studies of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10856_2015_5553_MOESM1_ESM.pdf
Supplementary material 1 (PDF 86 kb). Supplementary Materials in Online Ressource. Additional analyses of 1H → 31P CPMAS solid state NMR spectra (Figure S1)
Rights and permissions
About this article

Cite this article
Brouillet, F., Laurencin, D., Grossin, D. et al. Biomimetic apatite-based composite materials obtained by spark plasma sintering (SPS): physicochemical and mechanical characterizations. J Mater Sci: Mater Med 26, 223 (2015). https://doi.org/10.1007/s10856-015-5553-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10856-015-5553-9