
Abstract
Polycrystalline compounds La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) were elaborated using the ceramic method. All the samples are indexed in the orthorhombic structure (Pnma space group). Zero-field-cooled (ZFC) and field-cooled (FC) magnetization measurements show that the samples undergo a paramagnetic-ferromagnetic phase transition and the transition temperature is found to increase from 140.82 K for x = 0.00 to 191.50 K, for x = 0.20. The compounds reveal a second-ordered magnetic phase transition around TC. Isothermal entropy change \(|-\Delta {S}_{M}|\) was estimated using the Maxwell relation method. The relative cooling power also increases from 158.12 (J/Kg) for x = 0.00 to 175.94 (J/Kg) for x = 0.20, under a magnetic field of 5 T. The results suggest that materials could be useful for magnetic refrigeration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The development of new refrigeration technology, based on the magnetocaloric effect (MCE) of a magnetic solid, has a significant interest in recent years, as an efficient cooling technology and environmentally benign than the traditional gas compression refrigeration technology [1].
The MCE is quantified by the isothermal magnetic entropy change (\(\Delta {S}_{M}\)) or the related adiabatic temperature change (ΔTad) under an applied magnetic field. This phenomenon is originated from the coupling of the applied magnetic field with magnetic spins in a material [2, 3].
In most magnetic refrigeration prototypes, the lanthanide metal gadolinium (Gd) was the best room temperature magnetic cooling material with a Curie temperature is close to room temperature (TC = 294 K) and it possesses a large magnetic entropy change under a magnetic applied field of 5 T (\(-\Delta {S}_{M}\) = 10.3 J/(Kg K)) [4].
However, the high cost and easy oxidation of Gd rare earth metal limit their usage as an active magnetic refrigerant in magnetic refrigerators (MR). For these reasons, exploring new candidate materials for MR becomes necessary.
Taking this into consideration, several researchers groups focus on the search for cost-effective magnetic materials and have a magnetocaloric effect to apply as a magnetic refrigerator.
A large overview of the magnetocaloric effect observed in perovskite manganites, which makes them promising candidates for magnetic refrigeration. Moreover, these perovskite oxides are easy to prepare and show higher chemical stability. Another important feature of these compounds is the ability to control their physical properties by the type and the concentration of doping elements in A- and/or B -sites.
Among them, the manganites with the general system of Ln1−xAxMnO3 (where Ln = rare-earth elements and A = divalent alkali earth cations) show significant MCE around the TC [5,6,7,8,9].
In addition, this family of materials generally offers a high degree of chemical versatility and different other physical properties, such as colossal magnetoresistance [10, 11], superconductivity [12], spin-glass states [13], and phase separation [14], which find a wide range of other applications such as the spin-electronic devices and the magnetic field sensors. The magnetic properties in the perovskite manganites system have been explained by several theories, such as double exchange (DE) interaction [15], super-exchange (SE) coupling [16], and Jahn–Teller distortion [17]. Thus, the nature of the magnetic ordering with doped manganites depends on the relative concentration of the Mn3+ and Mn4+ ions, and the structural properties such as Mn–O bond length and Mn–O–Mn bond angle. Hence, the existence of the Mn3+–Mn4+ mixed-valence is necessary to introduce both ferromagnetic state and metallic conductivity.
Previously, has been reported that LaMnO3 is an antiferromagnetic (AFM) insulating compound [18]. It has been shown that the substitution of La3+ by a divalent alkali metal leads to a mixed-valence Mn3+/Mn4+ state and induces a transition from paramagnetic-insulator to ferromagnetic-metallic phase [19, 20].
Among the A site doped lanthanum manganites, the La1−xMgxMnO3 system can be more interesting as the doping can modify the lattice parameters and the Curie temperature of the magnetic phase transition. According to J.H Zhao et al. [21], the transition temperature of the system La1−xMgxMnO3 (0.05 ≤ x ≤ 0.4) decrease with increasing of the doping concentration (Mg content) from 147.2 K for x = 0.05 to 114.8 K for x = 0.2.
In addition, the average site cation radius ‹rA›, as well as the ratio of Mn3+/Mn4+ can be modified also by the creation of a vacancy in the A perovskite site. Recent studies on the lanthanum-deficiency effects in La0.7−xCa0.3MnO3 powder samples showed an increase in the Curie temperature TC with increasing deficiency content [22].
Traditionally, the solid-state reaction method has been shown as a very versatile technique to prepare solid solutions and other metastable systems. The principal objective of the traditional process is to elaborate a single phase of the compound. In the present work, we report the effect of La deficiency on structural, magnetic, and magnetocaloric properties of microcrystalline La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2).
2 Characterization
The samples La0.77−xMg0.23MnO3 were prepared by mixing the followed raw materials: La2O3, MgO, and MnO2, all powders used are from Sigma-Aldrich with a purity higher than 99.9%, in the desired proportion according to the following reaction:
The starting oxides are mixed in an agate mortar and then heated in air for 14 h at 900 °C. Next, the powders were then pressed into pellets (of about 2 mm thickness and 13 mm diameter under a pressure of 4 tons/in) and sintered at 1100 °C in the air for 36 h with intermediate grinding and pelleting cycles. Identification of the phase was investigated using a Panalytical X’pert PRO3 diffractometer with a Cu-Kα radiation.
(λ = 1.5406 Å). The obtained data were refined by the Rietveld refinement method with the FullProf program [23].
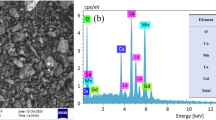
Scanning electron microscopy (SEM, TESCAN Vega 3SEM) was used to examine the morphology of our powder compounds.
Raman spectra were recorded on a Jobin Yvon HR 800 (λ = 441.6 nm) Raman spectroscopy at room temperature.
The FTIR (Fourier transformation infrared spectroscopy) spectrums of the studied materials were registered at room temperature using the FTIR Bruker Tensor 27 spectrometer in the 400–1200 cm−1wavenumber range.
ZFC and FC magnetization measurements vs. temperature (T) were carried out using a vibrating sample magnetometer (VSM, Cryogenic—Cryofree) in the temperature range of 10–350 K, under an applied field of 2000 Oe. Isothermal measurements M (H) were performed by varying H up to 5 T around the TC at several temperatures. The MCE as a magnetic entropy change was estimated from the magnetic isotherms for all samples.
3 Results and discussion
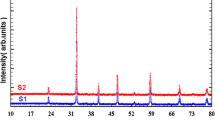
The room temperature X-ray diffraction (XRD) patterns of the La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) samples as well as the Miller index are present in Fig. 1.

X-ray diffraction patterns of La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) compounds at room temperature
All the experimental peaks are indexed in orthorhombic symmetry.
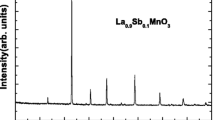
Figure 2 reports the refinement of XRD patterns for all the samples. It was found that our compounds crystallize in the orthorhombic structure with the Pnma space group (JCPDS data card No. 04-015-1752). For x = 0.2, the refinement has revealed the presence of a small impurity attributed to Mn3O4 phase. Essentially, it is necessary to note that the stoichiometry of the compound is not affected by the formation of the Mn3O4 secondary phase according to [24]. We can note that a lanthanum deficiency does not change the perovskite structure. Detailed results of the refinements are given in Table 1.

Rietveld refinement of the X-ray diffraction powder pattern of La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2)
Figure 3 shows that the unit cell volume decreases with increasing La deficiency amount. This structural change is caused by the increase of the Mn4+ content. Indeed, the Mn4+ ion has a lower ionic radius (< rMn4+ > = 0.53 Å) compared to that of (< rMn3+ > = 0.65 Å) Mn3+ [25].

Unit cell volume as a function of La-vacancy rate (x)
DIAMOND software is used for the estimation of the differences between the bonds Mn–O and the angles Mn–O–Mn [26]. From Table 2, it can be seen that the Mn–O length decreases while the Mn–O–Mn bond angle increases almost linearly with the La deficiency amount. Similar results are observed in various perovskite manganites [27, 28].
The average crystallite size (DSC) was calculated from the most intense XRD peaks using the Debye–Scherrer equation [29]:
where λ presents the X-ray wavelength (λ = 1.5405 Å), β the width at half maximum of the most intense peak and \(\theta\) the Bragg angle.
Crystallite size (DSSP) was obtained by the size–strain plot method according to the following relation [30]:
where dhkl represents the distance between (hkl) planes, k is the shape factor of the peak, and ε is the effective strain.
According to the above expression, a graph between \({({d}_{hkl}\beta \mathrm{cos}\theta )}^{2}\) against \(({d}_{hkl}^{2}\beta \mathrm{cos}\theta )\) is plotted and fitted linearly for all the compounds. The crystallite size is determined from the value of the slope of the linearly fitted data line (Fig. 4).

The size–strain plot (SSP) plots for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) compounds
The Halder–Wagner (H–W) analysis is another method to determine the crystallite size (DWH) using the following equation [31]:
where \(\beta^{*} = \frac{\beta \cos \theta }{\lambda },\,d^{*} = \frac{2\beta \sin \theta }{\lambda }.\)
A plot between \(\left( {\frac{{\beta_{{{\text{hkl}}}}^{*} }}{{d_{{{\text{hkl}}}}^{*} }}} \right)^{2}\) and \(\left( {\frac{{\beta_{{{\text{hkl}}}}^{*} }}{{d_{{{\text{hkl}}}}^{*} \,^{2} }}} \right)\) gives a straight line and the average crystal size have been obtained from the value of the slope of the line (Fig. 5).

The Halder–Wagner (H–W) plots for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) compounds
The extracted values of DSC, DSSP, and DWH are given in Table 3.
To describe the distribution of electron density (ED) of La0.77−xMg0.23MnO3 (x = 0.0, 0.05, 0.1, 0.15, and 0.2) compounds, the study of electron density mapping has been performed and delineated using the GFourier program in FullProf software. Two-dimensional maps distribution of Mn and O atoms along xz direction in all samples is displayed in Fig. 6. The dense and thick circular of the contours around Mn might be assigned to the distribution of valence d orbital electrons. Some intermediate positive contour of density is observed between the Mn cations correspond to the 2p sites for O2− cations. The reconstructed 2D electron density reveals the overlap between the density of Mn and O atoms, which is attributable to spin–orbit coupling interaction between the unpaired 2p core-level electrons and unpaired 3d valence shell electron in terms of Mn (3d eg)–O (2p) bonding bands. ED distribution around Mn–O bonds are found to be 1.113, 1.114, 1.185, 1.187, and 1.668 e/Å3 for x = 0, 0.05, 0.1, 0.15, and 0.2, respectively. Hence, can be seen that the electron density of Mn–O bands ameliorated slightly compared to the un-doped compound (x = 0.0). It is known that the electron density eventually affects the electrical and magnetic properties in these manganite systems. Thus, we expect that the magnetic properties of this material could be enhanced moderately [32].

Electron density map of La0.77−xMg0.23MnO3 (x = 0.0, 0.05, 0.1, 0.15, and 0.2) compounds
The SEM images and the grain size histogram plots are introduced in Fig. 7. SEM images reveal a non-uniform distribution of grains. The acquired grain size findings from SEM micrographs (using the Image J software by fitting the particle size distribution with a Gaussian equation) for x = 0.0, 0.05, 0.1, 0.15, and 0.2 are 1.1, 1, 0.9, 0.8, and 0.95 µm, respectively. They are much larger than those calculated by the Debye–Scherrer, the size–strain plot, and the Halder–Wagner (H–W) methods from XRD (Table 3), which indicate that each grain is observed by SEM consists of several crystallites.

Typical scanning electron micrographs and corresponding size distribution histograms of La0.77−xMg0.23MnO3 (x = 0.0, 0.05, 0.1, 0.15, and 0.2) samples
The infrared transmission spectra from 350 to 4000 cm−1 for all compounds measured at room temperature are presented in Fig. 8. The vibration band at 402.72 cm−1, observed in all samples, is attributed to the bending mode of Mn–O–Mn sensitive to the bond angle. The band located at 628.28 cm−1 corresponds to the stretching mode and is related to the change in length of the Mn–O–Mn or Mn–O bond [33, 34]. It is worth noticing that these two vibrations are related to the environment surrounding the MnO6 octahedra and the lowering of symmetry under the influence of a Jahn–Teller (JT) distortion. Remarkably, we show that for both bands no consistency changes with La-vacancy amount can be found.

FT-IR spectra for La0.77−xMg0.23MnO3 (0.0 ≤ x ≤ 0.2) samples
Figure 9 displays the zero-field-cooled (ZFC) and the field-cooled (FC) magnetization curves taken at 2000 Oe. It can be seen that the samples exhibit a paramagnetic to ferromagnetic transition with decreasing temperature. The differentiation curves (dM/dT) are shown in the inset of Fig. 9. These curves are refined by the Gauss equation to determine precisely the Curie temperature. With the increase in La deficiency, TC enhances considerably, from 140.82 K for x = 0 to 191.50 K for x = 0.2, which indicates the improvement of the ferromagnetism. These observations are consistent with the studies reported in the literature for other similar materials [35, 36]. Thereby, the increase of the Mn4+ ions produced by the La deficiency enhanced the double exchange interaction. This factor explains the increase of the Curie temperature in these studied materials.

Temperature dependence of ZFC and FC magnetization data taken at H = 2000 Oe for x = 0, 0.05, 0.1, 0.15, and 0.2. Inset: derivative of magnetization marking the ordering temperature
In addition, there is no bifurcation in the FC-ZFC M (T) modes; this asserts that the magnetic interactions are purely ferromagnetic at low temperatures.
The thermal evolution of the inverse of the susceptibility (χ−1) for all the samples is reported in Fig. 10. Thus, it is clear that the susceptibility obeys the Curie–Weiss law [37]:

Inverse magnetic susceptibility vs. temperature of the La0.77−xMg0.23MnO3 (x = 0, 0.05, 0.1, 0.15, and 0.2) compounds
where θp is the Curie Weiss temperature and C is the Curie constant.
The θp and C parameters, deduced from the linear fit in the paramagnetic region, are presented in Table 4. The values of the θp are positive and superior to the corresponding Curie temperatures, showing that the dominant interactions between Mn ions are ferromagnetic.
The Curie constant calculated from the fitting has been used to estimate the experimental effective magnetic moments \({\mu }_{eff}^{exp}\) by the equation C = NA \({\mu }_{B}^{2}\)/3kB \({(\mu }_{eff}^{2}\)) [38].
Where μB = 9.27 × 10–24 J.T−1 is the Bohr magneton and kB = 1.38 × 10–23 J.K−1 is the Boltzmann constant. Whereas the theoretical effective moments \({\mu }_{eff}^{th}\) are concluded from the genre formula of our present system, according to the following relation:
With \({\mu }_{eff}\)(Mn3+) = 4.9 μB and \({\mu }_{eff}\)(Mn4+) = 3.87 μB.
The values of \({\mu }_{eff}^{exp}\) and \({\mu }_{eff}^{th}\) are summarized in Table 4.
We observed there is a difference between the \({\mu }_{eff}^{exp}\) and \({\mu }_{eff}^{th}\) values, which can be related to the presence of ferromagnetic-spin interactions in the paramagnetic state [39].
Complementary, we have measured hysteresis loops between − 10 and 10 T at 5 K. The isothermal field-dependent magnetization loops curves are plotted in Fig. 11. The inset of Fig. 11 exhibits a zoom of the hysteresis loops at a low field for all the compounds.

The magnetization hysteresis curves of La0.77−xMg0.23MnO3 (x = 0, 0.05, 0.1, 0.15, and 0.2) samples measured at 5 K. The inset shows a zoom of the hysteresis loops at a low field for the samples
The compounds show a ferromagnetic behavior, we can also observe that magnetization gradually increases with the La deficiency (x). It is additionally seen that the magnetization increases sharply with the magnetic applied field and then saturates (x = 0.1 and x = 0.15). However, we show the absence of M (H) saturation for samples with x = 0, 0.05, and 0.2. This result confirms the presence of weak AFM interactions.
Isothermal magnetization curves have been undertaken under applied magnetic field ranging at 0–5 T for all samples and shown in Fig. 12. Is noticed that the magnetization increases regularly with increasing magnetic field and then saturates for fields above 1 T. A similar behavior was also depicted previously for La0.8Ba0.1Ca0.1Mn0.85Co0.15O3 [40] and La0.65Dy0.05Sr0.3MnO3 [41]. The nature of the magnetic phase transition can be deduced by plotting M2 versus H/M from the nature of the slope of the resulting curves according to Banerjee [42, 43]. Indeed, the magnetic transition is of the second-order type if the curves have a positive slope and of a first-order, if the slope is negative. As seen in Fig. 13, a positive slope and linear behavior of the Arrott plots, which indicates a second-order phase transition for all compounds.

Magnetization vs field isotherms near TC for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) samples

Arrott plots for La0.77−xMg0.23MnO3(0 ≤ x ≤ 0.2) samples
The change in magnetic entropy (\({\Delta S}_{M }\left(H,T\right)\)) of magnetic materials with second-order transitions can be calculated using the Maxwell thermodynamic relation [44]:
The magnetic entropy change between 0 and H (the maximal value of the applied magnetic field) defined in Eq. (7) could be approximated as [45]:
where \({\mathrm{M}}_{\mathrm{i}}\) and \({\mathrm{M}}_{\mathrm{i}+1}\) are the experimental values of the magnetizationat temperatures \({\mathrm{T}}_{\mathrm{i}}\) and \({\mathrm{T}}_{\mathrm{i}+1}\) under a magnetic field \({\Delta \mathrm{H}}_{\mathrm{i}}\).
Figure 14 displays the isothermal entropy \(-{\Delta S}_{M }\left(T\right)\) of the La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) compounds under several magnetic fields (from 1 to 5 T).

Thermal evolution of the entropy under different amplitudes of change in the magnetic field (from bottom to top ∆H = 1, 2, 3, 4, and 5 T) for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) samples
One notices clearly, that the \(-{\Delta S}_{M }\left(T\right)\) curves for all samples present a maximum close to the Curie temperature for each magnetic applied field. Also, the value of \(-{\Delta S}_{M}^{max}\) increases with increasing the applied magnetic field. The maximum values of the magnetic entropy change \(|-{\Delta S}_{M}^{max}|\), under 5 T magnetic field change, are (see the Fig. 15a and Table 4) 2.59, 2.65, 2.78, 2.40, and 2.60 J kg−1 for x = 0.0, 0.05, 0.1, 0.15, and 0.2, respectively. Comparable results were explored by R. Ben Hassine et al. [46] for La0.67Sr0.22Ba0.11Mn1-xFexO3 (x = 0, 0.1, and 0.2) compounds under the same applied magnetic field.

Variation of the maximum entropy change (a), the relative cooling power (b) as a function of the applied magnetic field for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) compounds
In addition to \(-{\Delta S}_{M}^{max}\), the relative cooling power (RCP) is another important parameter to evaluate the efficiency of magnetic refrigerators. The RCP can be calculated using the following relation [47]:
where \({\Delta T}_{FWHM}\) is the full width at half maximum of \(|-{\Delta S}_{M}^{max}|\)curve.
The RCP values obtained upon different magnetic applied field changes of μ0H = 1, 2, 3, 4, and 5 T, are displayed in Table 5 and Fig. 15b. We can see that the RCP values are improving with the increase of the La deficiency.
According to the thermodynamic theory, the change of the specific heat \({\Delta \mathrm{C}}_{\mathrm{p}}\) linked to a magnetic field variation can be calculated from the \(\Delta {S}_{M}\) data using the equation [48]:
The specific heat changes \({\Delta C}_{p}\) of the La0.77−xMg0.23MnO3 samples versus temperature at several magnetic fields is presented in Fig. 16.

Temperature dependence of the specific heat change at different applied magnetic field variations for La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2) samples
One can see clearly that the curves of \({\Delta C}_{p}\) indicate a change from a negative minimum \({\Delta C}_{P}^{min}\) to positive maximum \({\Delta C}_{P}^{max}\) values around the Curie temperature TC. Moreover, we find that the \({\Delta C}_{P}^{max}/{\Delta C}_{P}^{min}\) values increase with the applied magnetic field. These values and behavior are incoherent with those reported in the literature [50,51,52].
4 Conclusion
La0.77−xMg0.23MnO3 powder samples were prepared using the standard ceramic process for the composition range (0 ≤ x ≤ 0.2). X-ray diffraction shows that these perovskites crystallize in an orthorhombic structure (Pnma space group). Magnetic measurements show that all samples presented a second-order PM-FM, phase transition with an increase in Curie temperature TC. We have also mentioned the maximum values \(-{\Delta S}_{M}^{max}\) that varied between 2.40 and 2.78 J/KgK. Besides, the relative cooling power (RCP) increases from 158.12 (J/Kg) for x = 0.00 to 175.94 (J/Kg) for x = 0.20, under a magnetic field of 5 T, with increasing La deficiency. According to the results, La deficiency increases the cooling power, which could be a method to be considered as a promising way to develop new materials for magnetic refrigeration technology.
Data availability
All data generated or analyzed during this study are included in this published article.
References
C. Zimm, A. Jastrab, A. Sternberg, V. Pecharsky, K. Gschneidner Jr., M. Osborne, I. Anderson, J. Adv. Cryog. Eng. 43, 1759–1766 (1998)
S.M. Benford, G.V. Brown, T-S diagram for gadolinium near the Curie temperature. J. Appl. Phys. 52, 2110 (1981)
K.A. Gschneidner Jr., V.K. Pecharsky, A.O. Tsokol, Recent developments in magnetocaloric materials. J. Rep. Prog. Phys. 68, 1479 (2005)
SYu. Dankov, A.M. Tishin, V.K. Pecharsky, K.A. Gschneidner, Magnetic phasetransitions and the magnetothermal properties of gadolinium. J. Phys. Rev. B 57, 3478 (1998)
Y. Regaieg, M. Koubaa, W. Cheikhrouhou-Koubaa, A. Cheikhrouhou, T. Mhiri, J. Alloy. Comp. 502, 274 (2010)
V.B. Naik, R. Mahendiran, C. Bakya Jr., J. Appl. Phys. 110, 053915 (2011)
Y. Regaieg, M. Koubaa, W. Cheikhrouhou-Koubaa, A. Cheikhrouhou, L. Sicard, S. Ammar-Merah, F. Herbst, J. Mater. Chem. Phys. 132, 839 (2012)
F. Ayadi, W. Cheikhrouhou-Koubaa, M. Koubaa, S. Nowak, L. Sicard, S. Ammar, A. Cheikhrouhou, J. Mater. Chem. Phys. 145, 56 (2014)
Y. Regaieg, L. Sicard, J. Monnier, M. Koubaa, S. Ammar-Merah, A. Cheikhrouhou, J. Appl. Phys. 115, 17A917 (2014)
N. Abdelmoula, E. Dhahri, N. Fourati, L. Reversat, J. Alloy. Comp. 365, 25 (2004)
Y. Tokura, Y. Tomioka, J. Magn. Magn. Mater. 200, 1 (1999)
L. Laroussi, E. Dhahri, C. Boudaya, A. Cheikh-Rouhou, J. Ann. Chim. Sci. Mat. 23, 221 (1998)
M. Omri, M. Bejar, E. Sajieddine, E.K. Dhahri, M. Hlil, J. Es-Souni, J. Phys. B 407, 2566 (2012)
M.T. Tlili, N. Chihaoui, M. Bejar, E. Dhahri, M.A. Valente, E.K. Hlil, J. Alloy. Comp. 509, 6447 (2011)
Y.A. Izyumov, Y.N. Skryabin, Double exchange model and the unique properties of the manganites. J. Phys.-Usp. 44, 109 (2001)
J.M.D. Coey, M. Viret, S. Von Moln, Mixed-valence manganites. J. Adv. Phys. 48, 167–293 (1999)
A.J. Millis, P.B. Littlewood, B.I. Shraiman, J. Phys. Rev. Lett. 74, 5144 (1995)
S. Naji, A. Benyoussef, A. El Kenz, H. Ez-Zahraouy, M. Loulidi, J. Phys. A. 391, 3885–3894 (2012)
S. Lye, W.H. Song, J.M. Dai, K.Y. Wang, S.G. Wang, C.L. Zhang, J.J. Du, Y.P. Sun, J. Fang, J. Magn. Magn. Mater. 248, 26–33 (2002)
S. Das, A. Poddar, B. Roy, S. Giri, J. Alloy. Comp. 365, 94–101 (2004)
J.H. Zhao, H.P. Kunkel, X.Z. Zhou, G. Williams, J. Phys: Condens. Matter. 13, 9349–9367 (2001)
I. Kammoun, W. Boujelben, A. Cheikhrouhou, H. Roussel, R. Madar, J. Phys. Stat. Sol. C1, 1631 (2004)
J. Rodrigues-Carvajal, FULLPROF: A Rietveld Refinement and Pattern Matching Analysis Program (CEA-CNRS, France, 2000)
L.E. Heuso, P. Sande, D.R. Miguens, J. Rivas, F. Rivadulla, M.A. Lopez Quintela, J Appl Phys 91, 994 (2002)
D. Varshney, M.W. Shaikh, N. Dodiya, I. Mansuri, Structural properties of potassium doped lanthanum manganites. J. AIP Proc. 1349, 155 (2011)
G. Bergerhoff, M. Berndt, K. Brandenburg, Evaluation of crystallographic data with the program DIAMOND. J. Res. Natl. Inst. Stand. Technol. 101, 221–225 (1996)
W. Cheikhrouhou-Koubaa, M. Koubaa, A. Cheikhrouhou, W. Boujelben, A.-M. Haghiri-Gosnet, J. Alloy. Compd. 455, 67 (2008)
I. Walha, H. Ehrenberg, H. Fuess, A. Cheikhrouhou, J. Alloy. Compd. 485, 64–68 (2009)
S.K. Abdel-Aal, A.S. Abdel-Rahman, Graphene influence on the structure, magnetic, and optical properties of rare-earth perovskite. J. Nanopart. Res. 22, 1–10 (2020)
S.K. Abdel-Aal, A.I. Beskrovnyi, A.M. Ionov, R.N. Mozhchil, A.S. Abdel-Rahman, J. Phys. Status Solidi A 218, 2100138 (2021)
S.K. Abdel-Aal, M.F. Kandeel, A.F. El-Sherif, A.S. Abdel-Rahman, Synthesis, characterization and optical properties of new organic-inorganic hybrid perovskites [(NH3)2(CH2)3]CuCl4 and [(NH3)2(CH2)4] CuCl2Br2. J. Phys. Status Solidi A 218, 2100036 (2021)
I.Z. Al-Yahmadi, A. Gismelssed, I.A. Abdel-Latif, F. Al Ma’Mari, A. Al-Rawas, S. Al-Harthy, I.A. Al-Omari, A. Yousf, H. Widatallah, M. ElZain, T.Z. Myint, Giant magnetocaloric effect and magnetic properties of nanocomposites of manganite Nd1-xSrxMnO3 (0.0 ≤ x ≤ 0.8) synthesized using modified sol-gel method. J. Alloy. Compd. 857, 157566 (2020)
V.S. Kolat, H. Gencer, M. Gunes, S. Atalay, J. Mater. Sci. Eng. B 140, 212–217 (2007)
S. Keshri, L. Joshi, S.K. Rout, J. Alloy. Comp. 485, 501–506 (2009)
W. Boujelben, A. Cheikh-Rouhou, J.C. Joubert, J. Solid-State Chem. 156, 68–74 (2001)
W. Boujelben, A. Cheikh-Rouhou, H. Roussel, J. Magn. Magn. Mater. 290–291, 952–954 (2005)
J. Yang, Y.Q. Ma, R.L. Zhang, B.C. Zhao, R. Ang, W.H. Song, Y.P. Sun, J. Solid State Commun. 136, 268 (2005)
C. Bogdan, Synthèse et caractérisation de pérovskites doubles magnétorésistives dérivées de Sr2FeMoO6, Thèse, Université Paris Sud—Paris XI, 2004
B. Martinez, V. Laukhin, J. Fontcuberta, L. Pinsard, A. Revcolevschi, Magnetic field and pressure effects on the magnetic transitions of La0.9Ca0.1MnO3 perovskites. J. Phys. Rev. B 66, 056 (2002)
M.A. Gdaiem, S. Ghodhbane, A. Dhahri, J. Dhahri, E.K. Hlil, J. Alloy. Compd. 681, 547–554 (2016)
R. Felhi, M. Koubaa, W. Cheikhrouhou-Koubaa, A. Cheikhrouhou, J. Alloy Compd. 726, 1236–1245 (2017)
J. Mira, J. Rivas, F. Rivadulla, C. Vázquez-Vázquez, M.A. López-Quintela, J. Phys. Rev. B 60, 2998 (1999)
R. Skini, A. Omri, M. Khlifi, E. Dhahri, E.K. Hlil, J. Magn. Magn. Mater. 364, 5–10 (2014)
V.K. Pecharsky, K.A. Gschneidner Jr., Magnetocaloric effect and magnetic refrigeration. J. Magn. Magn. Mater. 200, 44–56 (1999)
M.H. Phan, S.C. Yu, Review of the magnetocaloric effect in manganite materials. J. Magn. Magn. Mater. 308, 325–340 (2007)
R. Ben Hassine, W. Cherif, J.A. Alonso, F. Mompean, M.T. Fernandez-Díaz, F. Elhalouani, J. Alloy. Compd. 649, 996–1006 (2015)
E. Sellami-Jmal, A. Marzouki-Ajmi, W. Cheikhrouhou-Koubaa, M. Koubaa, A. Cheikhrouhou, S. Nowak, L. Sicard, S. Ammar-Merah, N. Njah, J. Supercond. Novel Magn. 28, 2409–2415 (2014)
E. Dhahri, E.K. Dhahri, J. Hlil, J. RSC Adv. 9, 5530–5539 (2019)
C.P. Reshmi, S.S. Pillai, K.G. Suresh, M.R. Varma, Room temperature magnetocaloric properties of Ni substituted La0.67Sr0.33MnO3. J. Solid State Sci. 19, 130–135 (2013)
H. Yang, Y.H. Zhu, T. Xian, J.L. Jiang, Synthesis and magnetocaloric properties of La0.7Ca0.3MnO3 nanoparticles with different sizes. J. Alloy. Compd. 555, 150 (2013)
I. Messaoui, K. Riahi, W. Cheikhrouhou-Koubaa, M. Koubaa, A. Cheikhrouhou, E.K. Hlil, Phenomenological model of the magnetocaloric effect on Nd0.7Ca0.15Sr0.15MnO3 compound prepared by ball milling method. J. Ceram. Int. 42, 6825 (2016)
I. Messaoui, M. Kumaresavanji, K. Riahi, W. CheikhrouhouKoubaa, M. Koubaa, A. Cheikhrouhou, Investigation on magnetic and magnetocaloric properties in the Pb-doped manganites La0.78Ca0.22-xPbxMnO3 (x = 0, 0.05 and 0.1). J. Alloy. Compd. 693, 705 (2017)
Acknowledgements
This paper within the framework of collaboration is supported by the Tunisian Ministry of Higher Education and Scientific Research and the Portuguese Ministry of Science, Technology and Higher Education. The authors acknowledge the i3N (UID/CTM/50025/2020) and CICECO-Aveiro Institute of Materials (UID/CTM/50011/2020), financed by FCT/MEC and FEDER under the PT2020 Partnership Agreement. This work is also funded by national funds (OE), through FCT—Fundação para a Ciência e a Tecnologia, I.P., in the scope of the framework contract for eseen in the numbers 4, 5, and 6 of the article 23, of the Decree-Law 57/2016, of August 29, changed by Law 57/2017, of July 19.
Author information
Authors and Affiliations
Contributions
RS: preparing of samples, writing-original draft, revising. WC: investigation, revising. ARS: X-ray measurement, software. NMF: experimental measurements, investigation, revising. LK: investigation, supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Consent for publication
The authors declare that this manuscript has not been submitted to, nor is under review at, another journal or other publishing venue.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Selmi, R., Cherif, W., Sarabando, A.R. et al. Enhanced relative cooling power of lanthanum-deficiency manganites La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2): structural, magnetic and magnetocaloric properties. J Mater Sci: Mater Electron 33, 1703–1723 (2022). https://doi.org/10.1007/s10854-022-07726-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-022-07726-8