
Abstract
Tungsten oxide (WO3) nanoparticles were synthesized by microwave irradiation method in the presence of cetyltrimethylammonium bromide (CTAB) as a surfactant. The structural, morphological, optical and porous properties were studied. The X-ray diffraction pattern and Raman spectroscopy show that both the sample crystallizes in stable monoclinic structure with nanoplates morphology which is inferred from the image of high-resolution scanning electron microscopy. The use of CTAB as the surfactant decreased the optical band gap of WO3 from 3.27 to 3.24 eV. In addition to which the electrochemical measurements, cyclic voltammetry, chronopotentiometry and electrochemical impedance spectroscopy have been recorded. It is found that the surfactant-mediated WO3 has larger specific surface area, specific capacitance and energy density of 135.90 m2 g−1, 393 F g−1 and 6.69 Wh kg−1, respectively, than the pure WO3. The experimental results revealed that the use of CTAB as surfactant in tungsten oxide synthesis enhanced its electrochemical performance, and thus, it might be a suitable positive electrode material for supercapacitor application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The energy storage devices such as fuel cells, batteries and supercapacitors play a major role in coping up with the futuristic energy demand. Supercapacitors do not require continuous flow of external source as in fuel cell and has high power density than the batteries which made them to act as an efficient energy storage device. Among its types, asymmetric supercapacitors (ASCs) have gained greater attention in the recent years because of its high specific capacitance and cyclic stability in which efficiency is determined by the usage of suitable positive electrode material [1]. Recently, it is observed that high specific capacitance can be achieved by using transition metal oxides (TMOs), conducting polymers and MXenes as positive electrode material in ASCs. Of which TMOs play a major role as its faster redox reaction rate leads to higher specific capacitance and energy density [2].
Although TMOs such as RuO2, NiO, MnO2, V2O5 and Fe2O3 exhibit attractive performance, its criticality and toxicity act as a hurdle in its commercialization. High thermal stability, natural abundance, notable compatibility, low cost, high theoretical capacitance and wide negative operating potential window (~ 0.8 V) in aqueous electrolyte of tungsten oxide (WO3) hindered us to study its electrochemical performance [3]. h-WO3 synthesized on carbon cloth exhibits a gravimetric capacitance of 373 F g−1 at 1 A g−1 [4]. The metallic nanofilament array embedded WO3 was reported to have specific capacity of 214 F g−1 at 0.25 mA cm−2 [5]. The 2D h-WO3/Ti3C2 Mxene hybrid architectures were found to possess specific capacitance of 566 F g−1 [6]. Amit Kumar Das et al. [7] fabricated a flexible ASC device with NiSe and WO3@PPy composite as positive and negative electrodes, respectively, which exhibits promising energy density of 37.3 Wh kg−1 at 2 A g−1. Similarly, Pragati A. Shinde et al. [8] designed a flexible all solid state ASC with MWCNT-WO3 and MnO2 as negative and positive electrodes which achieve a specific capacitance of 145.6 F g−1 at a current of 2 mA. It is also known from the recent study that tuning of surface area in TMOs will result in enhanced electrochemical performance which can be done by using additives and nano structuring [9, 10]. Backing to the argument further, it is observed from Fig. 1 that the use of surfactant can improve the electrochemical performance of the active material [11,12,13,14,15,16,17].

Comparison of specific capacitance of various TMOs with and without surfactant
In this article, we intend to study the capability of nanostructured WO3 and the influence of surfactant in its electrochemical performance when it is used as a positive electrode material in supercapacitor application. In the present study, cetyltrimethylammonium bromide (CTAB), a positively charged cationic surfactant, is chosen as it plays a great role in tuning the nucleation and growth of nanostructured WO3 [18].
2 Materials and methods
Tungstic acid (H2WO4) and CTAB were used as precursor and surfactant, respectively. Nickel foam, carbon black, polyvinylidene difluoride (PVDF), and N-methyl-2-pyrolidinone were used for working electrode preparation.
Pure and CTAB-mediated tungsten oxide nanoparticles were synthesized by microwave irradiation method in a fashion similar to our previous work [19]. The detailed method of preparation is as follows. Initially, 2.49 g of tungstic acid is dissolved in 10 mL of NaOH solution and stirred for 30 min which ended up in the formation of hydrated sodium tungstate solution by proton exchange protocol. By adding HCl, the pH of the solution was adjusted to 1. Then the solution was subjected to microwave irradiation with a power of 240 W for 7 min. After that it was centrifuged several times at 5000 rpm with distilled water and ethanol. The obtained sample was dried at 60 °C for 12 h. This resulted in hydrated tungsten oxide which was annealed at 500 °C for 5 h to remove the water molecules and recrystallize to form WO3. To prepare CTAB-mediated WO3, after forming hydrated sodium tungstate solution as done earlier, 0.5 g of CTAB was added and stirred well for about 30 min. Later on, the same procedure was adopted as above. The final products of pure and CTAB-mediated WO3 have been named as S1 and S2 hereafter. The schematic of the synthesis procedure is shown in Fig. 2.

Systematic procedure for the synthesis of pure and CTAB-mediated WO3
The powder X-ray diffraction (XRD) measurements of the samples were taken from D8 ADVANCE X-ray diffractometer in DAVINCI DESIGN operated in Bragg–Brentano geometry with Cu-K-alpha radiation (λ = 1.5406 Å) as source for X-ray. With the help of Thermoscientific Apreo S High-Resolution Scanning Electron Microscope (HR-SEM), the morphology and elemental composition of the samples were studied. SHIMADZU’s UV 3600 plus spectrophotometer was used to record the Diffuse Reflectance Spectrum (DRS) in UV–Vis wavelength range. LabRAM HR evolution high spectral resolution confocal Raman microscope manufactured by HORIBA aids in observing the vibrating modes of the samples (Imaging Laser-532 nm). Nitrogen absorption/desorption equipment from Quantachrome Instruments (Autosorb IQ series) was used for surface area analysis.
In order to prepare working electrode, active material, carbon black and PVDF were taken in 85:10:5 ratios. First, carbon black and PVDF were grinded up for 30 min. Second, the active material was added to this mixture and ground again for 30 min. By adding N-methyl-2-pyrolidinone, this powder was made as slurry. This slurry was coated on the nickel foam (1 × 1 cm) which was ultrasonicated in ethanol already for about 30 min. Finally, this electrode was dried at 60 °C for 24 h and used as working electrode for electrochemical measurements. The mass loading was found to be 4 mg and 3.5 mg for S1 and S2 samples, respectively. The electrochemical studies were carried out in SP-300 model electrochemical workstation distributed by BIO-LOGIC SAS. In the typical experiment, 1 M KOH, Ag/AgCl, platinum wire and active material coated on nickel foam were used as electrolyte, reference, counter and working electrode, respectively, in three electrode system model.
3 Results and discussion
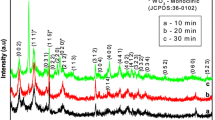
The XRD pattern of both S1 and S2 is shown in Fig. 3. The obtained patterns show major peaks at 23.6°, 24.4°, 33.4°, 34.1° and 50.15° corresponding to (020), (200), (022), (220) and (400) planes, respectively, indicating the stable monoclinic structure and match well with the JCPDS: 24-0747. The lattice parameters, average crystallite size (D), micro-strain (ε) and dislocation density (δ) of both the samples were calculated using the relations given below and the obtained values are recorded in Table 1.

XRD patterns of S1 and S2 samples
where λ—wavelength of the X-ray, β—full-width half maximum, K—Scherrer constant, and θ is the peak position [20,21,22]. Since the exact value of K is not known for the present system, K = 0.9 was used and the calculations of D based on this equation are estimates only. It is noted from Table 1 that the use of surfactant increases the crystallite size and decreases the dislocation density and micro-strain as surfactant induces the nucleation and growth of the particles.
The elmental composition of the synthesized samples was studied using Energy-Dispersive X-ray spectroscopy (EDX) and is shown in Fig. 4a, b. It is clear from the study that the W:O ratio was found to be 1:3 for both pure and surfactant-mediated WO3. Stochiometric WO3 is expected to crystalize in stable monoclinic structure which stands with the XRD analysis [3]. The morphology of the samples was studied by HR-SEM and is shown in Fig. 4c, d. It is observed from the image that S1 and S2 have heterogeneous and homogeneous nanoplates morphology, respectively. The average diameters of nanoplates of both the samples were around 34 nm.

a, b EDS of S1 & S2, c, d HR-SEM of S1 & S2
The mechanism involved in the formation of nanoplate morphology is clearly shown in Fig. 5. During the formation process, tungstic acid decomposes to form nuclei of WO3 which has microlayers of octahedrons [WO6]6−. In the absence of surfactant, these octahedrons in monoclinic structured WO3 will interact and grow in all directions resulting in the formation of heterogeneous nanoplates morphology for S1, whereas for the sample with surfactant, the ionized CTA+ being a cationic surfactant readily attaches to the four oxygen atoms and forms [CTA-WO6]2− by stereochemical effect. The attachment of CTAB to the nuclei constrains the growth along ‘a’ direction resulting in the homogeneous nanoplate morphology of S2 [23, 24].

Growth mechanism of pure and CTAB-mediated WO3
The phase and fundamental vibration modes of S1 and S2 were analysed using Raman spectroscopy, and it is shown in Fig. 6. The strongest wavenumbers were observed around 134, 272, 716 and 807 cm−1 for S1 and at 134, 273, 717 and 807 cm−1 for S2. The phonon contribution is low in 200–400 cm−1 range and is high in 600–900 cm−1. The presence of peak at 617 and 781 cm−1 indicates the WO2 phase. Since these peaks are absent, it is understood indirectly that both the samples crystallize in WO3 phase [25]. Raman peaks around 270, 330 cm−1 and 715, 807 cm−1 correspond to the bending and stretching vibration modes of O–W–O bonds in monoclinic WO3, respectively, which agree well with XRD [26].

Raman analysis of S1 and S2 samples
The band-gap evaluations of both the samples were done by recording diffuse reflectance spectra, shown in Fig. 7. The optical band gap of semiconductors can be calculated using the Tauc method. Relation between energy-dependent absorption coefficient (α) and energy gap (Eg) based on the assumptions from the Tauc method is expressed as follows:

UV-DRS spectra of S1 and S2 samples
where h—Planck constant, ν—phonon’s frequency, n—nature of electron transition (n = ½ and 2 for direct and indirect transitions, respectively) and B is a constant. To convert the reflectance data obtained from DRS to absorbance, Kubelka–Munk function can be used [27].
By incorporating (5) in (4), band-gap energy can be calculated which is done graphically as shown in Fig. 7. It is observed that the surfactant-mediated WO3 nanoparticles have smaller band gap as it is larger in size than the pure WO3 nanoparticles synthesized without CTAB. This smaller band gap promotes the rate of charge transfer in the electrode material.
In order to get a complete picture of the pore size distribution and surface area of the active material, the nitrogen adsorption and desorption analysis have been performed. The absorption and desorption plot are shown in Fig. 8. The isotherm of both the samples falls under the Type-IV of the IUPAC classification of physisorption. The hysteresis loop of S1 and S2 corresponds to the capillary condensation in mesopores. From the obtained adsorption data, the specific surface area and the pore size distribution (PSD) are calculated by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) method. Relative pressure P/P0 vs 1/[W ((Po/P) − 1)] within 0.05–0.30 (P/P0 range) gives the BET plot whose linear fit is used to calculate the specific surface area of the material given by the relation:

N2 adsorption–desorption isotherm of S1 and S2 samples
where \({n}_{m}^{a}\), L, am and m is the monolayer absorbance capacity, Avogadro number, molecular cross-sectional area and mass of the adsorbent, respectively. The specific surface area of S2 (135.90 m2 g−1) is higher than S1 (113.39 m2 g−1). The mesoporous structure (pore width 2–50 nm) and larger BET surface area of S2 help to enhance charge transfer and power capability by providing low resistance pathway [11]. The PSD of the samples is shown in the inset of Fig. 8 from which the pore radius and pore volume were estimated as 15.47 Å and 0.19 cm3 g−1 for S1 and 16.17 Å and 0.23 cm3 g−1 for S2, respectively [28, 29].
The Cyclic Voltammetry (CV) of both the samples S1 and S2 was recorded by applying the potential from 0 to 0.5 V at various scan rates of 5, 10, 20, 40, 60, 80 and 100 mV s−1 and is shown in Fig. 9a, b. The plot shows the peaks corresponding to the oxidation and reduction process indicating the pseudocapacitive behaviour of the material. The redox reaction [30] taking place at the interface is given as follows:

a, b CV plot and c charge storage mechanism in S1 and S2
The area enclosed by the curve for the scan rate of 100 mV s−1 is higher for the sample S2 (3.25 AV) than S1 (2.62 AV) which results in the higher specific capacitance of sample S2. The type of electrochemical energy storage mechanism (capacitive or semi-infinite diffusion) taking place within the active material can be estimated in the following way: the variation of current with scan rate can be mathematically represented as follows:
where i—current, v—scan rate, and a and b are constants. If b = 0.5, the charge storage is due to the semi-infinite diffusion and if b = 1, it is capacitive. The slope of the plot between log (Scan rate) and log (peak current) will result in b value (see Fig. 9c) [31]. The b value for the anodic and cathodic peaks of both S1 and S2 has been calculated and is shown in inset of Fig. 9c. It is seen that b value falls around 0.7 which indicates that the charge is stored in the active material by semi-infinite diffusion and capacitive as well.
The charging and discharging taking place at the interface of active material and electrolyte have been studied using the chronopotentiometry (CP) analysis. The charge and discharge mechanisms have been recorded for various current densities of 0.5–10 A g−1 and are shown in Fig. 10a, b. The CP plot has two regions indicating the charging and discharging due to the redox reaction conformed from the CV analysis which is the typical behaviour of the pseudocapacitive material. The drop at the starting of charging and discharging cycle is known as IR drop which is due to the ohmic resistance. It is also noted from the figure that the time taken for the process reduces drastically as we increase the current density. This reduction affects the magnitude of specific capacitance calculated using the relation:

a, b Charge/discharge plot of S1 and S2 and c variation of specific capacitance as a function of current density in S1 and S2
where i is the discharge current, ∆t is the time taken for complete discharge, m is the active mass of the material coated on the nickel foam and ∆V is the potential window [4]. The calculated specific capacitances for both S1 and S2 have been tabulated in Table 2. The Cp is found to decrease with the increase in current density (Fig. 10c). As indicated by the larger sweep area in CV and larger surface area in BET analysis, the specific capacitance of the sample S2 (393 F g−1) is higher than S1 (199 F g−1) at 0.5 A g−1.
The electrochemical impedance spectroscopy (EIS) was recorded for both the samples S1 and S2 in the frequency range of 1 Hz to 10 kHz in open-circuit potential mode which is shown in Fig. 11. The amplitude of the potential was given as 5 mV where the Z′ and Z′′ represent the real and imaginary parts of the impedance, respectively. It is observed from the figure that the Warburg impedance in the low-frequency region is steeper for S2 than S1. This implies that the sample S2 has low diffusive resistance than S1. Hence, the charge transfer between the active material and electrolyte is faster for WO3 with surfactant than pure WO3 [16].

Nyquist plot of S1 and S2 samples
The efficiency of the electrode material and its applicability are determined by the energy and power density which are plotted against each other using the Ragone plot. The energy and power density can be calculated from the chronopotentiometry data using the equations:
where C is the specific capacitance at a particular current density, V is the potential window, t is the discharge time, and the constants 3.6 and 3600 were included in the equations for the unit conversion [9]. The calculated E and P values for S1 and S2 are shown in Table 2 and their Ragone plots are compared in Fig. 12. It is obvious that the energy density of the electrode with surfactant is twice that of the one without surfactant.

Ragone plot of WO3 without and with surfactant
4 Conclusion
In summary, pure and CTAB-mediated WO3 nanoparticles have been synthesized by microwave irradiation method. The XRD analysis revealed that both the samples have been crystallized in the stable monoclinic structure. The samples S1 and S2 have heterogeneous and homogeneous nanoplate morphologies. It is observed that the presence of surfactant plays a major role in tuning the morphology of the sample. The stretching and bending vibrational modes of O–W–O have been observed in Raman analysis. The charge transfer rate is increased by the lower band gap of S2. BET analysis showed that WO3 with surfactant is found to have larger surface area of 135.90 m2 g−1 which enhances the specific capacitance of the active material. The electrochemical examination shows that the specific capacitance of S1 and S2 is 199 and 393 F g−1 at 0.5 A g−1, respectively. The energy density of the active material is doubled by adding the surfactant into it. These results show that the addition of surfactant (CTAB) during the synthesis of WO3 enhances the electrochemical performance of the material. We suggest that the use of WO3 with CTAB as a positive electrode material in ASCs will be a promising candidate for pseudocapacitive applications.
Data availability
The data and materials that support our published claims stand well with the field standards and Journal policies.
Code availability
Not Applicable.
References
Y. Shao, M.F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn, R.B. Kaner, Chem. Rev. (2018). https://doi.org/10.1021/acs.chemrev.8b00252
K. Poonam, A. Sharma, S.K. Arora, J. Tripathi, Energy Storage (2019). https://doi.org/10.1016/j.est.2019.01.010
P.A. Shinde, S.C. Jun, Chemsuschem (2020). https://doi.org/10.1002/cssc.201902071
T. Wang, H. Liu, J. Li, H.A. Munir, ChemistrySelect (2020). https://doi.org/10.1002/slct.202001733
J. Kim, A.I. Inamdar, Y. Jo, S. Cho, A.T.A. Ahmed, B. Hou, S. Cha, T.G. Kim, H. Kim, H. Im, J. Mater. Chem. A (2020). https://doi.org/10.1039/D0TA01728K
S.B. Ambade, R.B. Ambade, W. Eom, S.H. Noh, S.H. Kim, T.H. Han, Adv. Mater. Interfaces (2018). https://doi.org/10.1002/admi.201801361
A.K. Das, S. Paria, A. Maitra, L. Halder, A. Bera, R. Bera, S.K. Si, A. De, S. Ojha, S. Bera, S.K. Karan, B.B. Khatua, ACS Appl. Electron. Mater. (2019). https://doi.org/10.1021/acsaelm.9b00164
P.A. Shinde, Y. Seo, C. Ray, S.C. Jun, Electrochim. Acta (2019). https://doi.org/10.1016/j.electacta.2019.03.159
A. Biswal, P.K. Panda, A.N. Acharya, S. Mohapatra, N. Swain, B.C. Tripathy, Z. Jiang, M.M. Sundaram, ACS Omega (2020). https://doi.org/10.1021/acsomega.9b03657
K.K. Upadhyay, M. Altomare, S. Eugenio, P. Schmuki, T.M. Silva, M.F. Montemor, Electrochim. Acta (2017). https://doi.org/10.1016/j.electacta.2017.02.131
A. Juliet Christiana Mary, A. Chandra Bose, Appl. Surf. Sci. (2018). https://doi.org/10.1016/j.apsusc.2018.01.117
H. Zhang, Y. Wang, C. Liu, H. Jiang, J. Alloys Compd. (2012). https://doi.org/10.1016/j.jallcom.2011.11.042
S. Suthakaran, S. Dhanapandiyan, N. Krishnakumar, N. Ponpandian, P. Dhamodharan, M. Anandan, Mater. Sci. Semicond. Process. (2020). https://doi.org/10.1016/j.mssp.2020.104982
S. Asaithambi, P. Sakthivel, M. Karuppaiah, G. Udhaya Sankar, K. Balamurugan, R. Yuvakkumar, M. Thabidurai, G. Ravi, J. Energy Storage (2020). https://doi.org/10.1016/j.est.2020.101530
B. Saravanakumar, G. Ravi, V. Ganesh, F. Ameen, A. Al-Sabri, R. Yuvakkumar, J. Solgel Sci. Technol. (2018). https://doi.org/10.1007/s10971-018-4685-z
X. Zhang, Y. Zhao, C. Xu, Nanoscale (2014). https://doi.org/10.1039/C3NR06734C
A. Qian, K. Zhuo, M.S. Shin, W.W. Chun, B.N. Choi, C. Chung, Chemsuschem (2015). https://doi.org/10.1002/cssc.201403477
D. Sanchez-Martineza, C. Gomez-Solisa, L.M. Torres-Martineza, Mater. Res. Bull. (2015). https://doi.org/10.1016/j.materresbull.2014.10.034
S. Paulraj, R. Jayavel, J. Mater. Sci. Mater. Electron. (2018). https://doi.org/10.1007/s10854-018-9510-5
P. Scherrer, Nachr. Ges. Wiss. Göttingen 26, 98 (1918)
J.I. Langford, A.J.C. Wilson, J. Appl. Cryst. (1978). https://doi.org/10.1107/S0021889878012844
V. Uvarov, I. Popov, Mater. Charact. (2013). https://doi.org/10.1016/j.matchar.2013.09.002
D. Meng, G. Wang, X. San, Y. Shen, G. Zhao, Y. Zhang, F. Meng, J. Nanomater. (2015). https://doi.org/10.1155/2015/393205
R. Ponnusamy, A. Gangan, B. Chakraborthy, C.S. Rout, J. Appl. Phys. (2018). https://doi.org/10.1063/1.5010826
V.B. Kumar, D. Mohanta, Bull. Mater. Sci. (2011). https://doi.org/10.1007/s12034-011-0117-1
Y. Baek, K. Yong, J. Phys. Chem. C (2007). https://doi.org/10.1021/jp0659857
P. Makula, M. Pacia, W. Macyk, J. Phys. Chem. Lett. (2018). https://doi.org/10.1021/acs.jpclett.8b02892
K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquerol, T. Siemieniewska, Pure Appl. Chem. (1985). https://doi.org/10.1351/pac198557040603
Aashima, S. Uppal, A. Arora, S. Gautam, S. Singh, R.J. Choudhary, S.K. Mehta, RSC Adv. (2019). https://doi.org/10.1039/c9ra03252e
X. Cui, J. Shi, H. Chen, L. Zhang, L. Guo, J. Gao, J. Li, J. Phys. Chem. B (2008). https://doi.org/10.1021/jp803565k
M. Zhu, W. Meng, Y. Huang, Y. Huang, C. Zhi, A.C.S. Appl, Mater. Interfaces (2014). https://doi.org/10.1021/am504756u
Acknowledgements
The authors thank the SRM Institute of Science and Technology for providing High-Resolution scanning Electron Microscope (HR-SEM) and Micro-Raman Facility. We also acknowledge the Nanotechnology Research Centre (NRC), SRMIST for providing the Research Facility. We also gratefully acknowledge Prof. C. Muthamizhchelvan, Vice-Chancellor, and Prof. D. John Thiruvadigal, Chairperson (School of applied sciences) for the extended facilities.
Funding
No funding was received to conduct this study.
Author information
Authors and Affiliations
Contributions
VK: designed the work and worked out all the technical issue throughout the study. SP: designed the work and interpretation of the results. MK & SR: carried out the synthesis and experiments and also wrote the manuscript with the input from all the authors. VP and PV: revised the manuscript critically for important intellectual concepts. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kowsalya, M., Paulraj, S., Rajagopal, S. et al. Effect of surfactant on the electrochemical performance of WO3 as supercapacitor electrode. J Mater Sci: Mater Electron 33, 8833–8843 (2022). https://doi.org/10.1007/s10854-021-06915-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06915-1