
Abstract
In this work, a novel phenanthro[9,10-d]imidazole-zinc coordination complex (Zn(TPPI)2) is reported. This compound exhibits suitable physical properties for a host material. First, its highest occupied molecular orbital (HOMO) (− 5.33 eV) energy level is favorable for hole injection from transporting layer. Second, its small singlet–triplet splitting (0.31 eV) can reduce the intrinsic energy loss from singlet to triplet excited state and promote the device efficiency. Finally, its good thermal stability (decomposition temperature: 495 °C) favors evaporation preparation of uniform films. As a result, yellow–red and red phosphorescent devices which utilize Zn(TPPI)2 as host display high efficiencies (yellow–red: 58.35 cd/A, 70.52 lm/W, 20.82%) (red: 15.89 cd/A, 16.09 lm/W and 19.05%). The performance of red device is comparable to the best results of previous publications on corresponding emitter. Meanwhile, the undoped device of Zn(TPPI)2 exhibits high efficiencies (3.79 cd/A, 3.97 lm/W, 2.47%) in zinc-complex blue emitters. These results demonstrate that Zn(TPPI)2 is a good blue emitter and host material.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metal complex is widely used in organic light emitting diodes (OLEDs) [1,2,3]. Based on precious metals (iridium, platinum, palladium, gold, etc.), a large number of phosphorescent emitters have been developed and an ideal internal quantum efficiency of 100% is realized [4,5,6]. At present, phosphorescent metal complex has been adopted by practical application. Compared with precious metal complex, complex based on cheap metal (aluminium, lithium, zinc, copper, etc.) also gains much attention because of its much lower cost [7,8,9]. In these cheap metal complexes, zinc complex is an important kind [10,11,12]. Zinc complex possesses many advantages, such as excellent thermal stability, good charge-transporting ability, colorless product, low cost and easy preparation. Motivated by these fascinating characteristics, in the past decades, researchers have developed many zinc complexes for OLED and a large number of articles have been published [13,14,15,16,17]. These works demonstrate that zinc complex can be used as emitter, host and electron-transporting material [18,19,20,21,22]. Although the performance of zinc complex has been dramatically promoted, some aspects still need to be improved. First, the device efficiency of zinc-complex emitter is low, especially for blue emitter. At present, the efficiencies of reported zinc-complex blue emitters are less than 2.7 cd/A [23,24,25]. Second, few zinc-complex host exhibits comparable device performance to those pure organic hosts [26, 27]. Hence, to realize the practical application of zinc complex in OLEDs, developing novel high-performance zinc complex is still highly desired.
Herein, we utilized 2-(1,6,9-triphenyl-1H-phenanthro[9,10-d] imidazol-2-yl)phenol (TPPI) as ligand and developed a novel zinc complex (Zn(TPPI)2) (Scheme 1). Zn(TPPI)2 exhibited blue emission in both solution (467, 473 nm) and film state (447 nm). Its undoped blue device displayed high efficiencies (3.79 cd/A, 3.97 lm/W, 2.47%) in zinc-complex blue emitters [23, 25]. Meanwhile, the thermal stability and energy level of Zn(TPPI)2 met the requirements of an excellent host. Hence, yellow–red and red phosphorescent devices which utilized Zn(TPPI)2 as host exhibited high performance. These results demonstrated that Zn(TPPI)2 was a good blue emitter and host material.

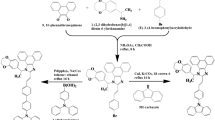
The synthetic procedures of Zn(TPPI)2
2 Experimental
2.1 General information
All the starting chemicals for the synthesis were purchased from commercial sources and used as received without purification unless otherwise noted. Dichloromethane and toluene for the third and fourth preparation steps were dried by sodium or calcium hydride and distilled under nitrogen atmosphere. 1H and 13C NMR spectra were measured using an AVANCE 600 MHz spectrometer (Bruker). Mass spectrometry were characterized by Shimazu LCMS2020 instrument. MALDI-TOF-MS was measured by Bruker Autoflex lll (Maldi-tof-MS) at Ceshigo Research Service. Elemental analyses were performed on a flash EA 1112 spectrometer. The contents of element carbon, hydrogen, nitrogen were measured directly and the content of oxygen was calculated. Ultraviolet–visible (UV–Vis) absorption spectra were tested using a Hitachi U-3900 spectrophotometer. A Horiba FluoroMax-4 spectrophotometer was used for fluorescence spectra. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) characteristics were determined by TA Q600 thermogravimeter and NETZSCH DSC204 instrument using the heating rate of 10 °C min−1 under nitrogen atmosphere to obtain the compound weight loss. The AFM image was measured by Dimension ICON (Bruker) at Ceshigo Research Service. The electrochemical properties were investigated by electrochemical workstation, the working electrode was a platinum electrode, the reference electrode was an Ag electrode, and the counter electrode was a platinum wire. The redox potential was performed in a 0.1 M solution at a scan rate of 100 mV s−1 with tetrabutylammonium hexafluorophosphate (TBAPF6) as the electrolyte and ferrocene (Fc) was added to calibrate the zero point after sample measurement finished. For reversible curves, the potential value was the average value of two reversible peaks (E = (E1 + E2)/2). For irreversible curves, at the onset of the first wave, two tangents were drawn and the intersection-point value was the potential value. To obtain the actual sample potential value (Esample), the sample measured value (Emeasure) should subtract the ferrocene measured value (EFc) (Esample = Emeasure − EFc). Then, from the oxidation and reduction peaks, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of sample were calculated according to the equation of (EHOMO/LUMO = − (E[onset, ox/red vs Fc+/Fc] + 4.8) eV. Finally, the energy gap (Eg) was the difference of HOMO and LUMO (Eg = LUMO–HOMO). The photoluminescence quantum yields (PLQYs) were measured with the Yokohama C9920-02G assay system.
The vacuum sublimation was carried out at the sublimation machine from Shenyang Keyou Vacuum Technology Co., Ltd. This machine had two heating sections to set up different temperatures. For Zn(TPPI)2, the two section temperatures were set to 260 and 370 °C, respectively. The vacuum degree was 31 Pa. After 12 h, the sublimation finished and pure Zn(TPPI)2 was obtained.
2.2 OLED fabrication and characterization
All the devices were fabricated on indium tin oxide (ITO) substrate with a sheet resistance of 15 Ω/sq and a thickness of 135 nm. The ITO substrate was cleaned with acetone, detergent, distilled water in sequence, and then in an ultrasonic solvent bath. After dried at 80 °C for 1 h, the ITO substrates were treated with UV-ozone for 15 min. The substrate was transferred to a vacuum deposition chamber. Dipyrazino [2,3-f:2′,3′-h] quinoxaline-2,3,6,7,10,11-hexacarbonitrile (HATCN), 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC), 4,4′,4″-tris (carbazol-9-yl)-triphenylamine (TCTA), Zn(TPPI)2, host–guest doped luminescent layer (Zn(TPPI)2: bis(2-phenyl-benzothiazole-C2, N)(acetylacetonate)iridium(III) (Ir(bt)2(acac)) or (Zn(TPPI)2: tris(1-phenylisoquinoline)iridium(III) (Ir(piq)3) or N, N-dicarbazolyl-3,5-benzene (mCP): Ir(bt)2(acac), 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB), lithium fluoride (LiF) and aluminium (Al) were evaporated sequentially through a shadow mask with an array of 0.1 cm2 at the pressure of 5.0 × 10–4 Pa. The current–voltage-luminance (J-V-L) characteristics were tested using a Keithley 2400 semiconductor characterization system and a calibrated silicon photodiode. The electroluminescent (EL) spectra and brightness were measured with spectrograph PR-670. All the measurements were carried out under argon conditions at room temperature.
2.3 Synthesis 6,9-dibromo-2-(2-methoxyphenyl)-1-phenyl-1H-phenanthro[9,10-d]imidazole (M1)
To a two-necks round-bottom flask, 3,6-dibromophenanthrene-9,10-dione (3.64 g, 10 mmol), aniline (3.72 g, 40 mmol), 2-methoxybenzaldehyde (1.36 g, 10 mmol), ammonium acetate (3.85 g, 50 mmol) and acetic acid (60 mL) were added. Under nitrogen atmosphere, the mixture was refluxed for 10 h. After cooling to room temperature, the solvent was removed by rotary evaporation and distilled water (100 mL) was added. The mixture was extracted by dichloromethane for three times (100 mL × 3) and then dichloromethane was removed by rotary evaporation to obtain the crude product. The crude product was purified by column chromatography on silica gel (eluent: dichloromethane/ethyl acetate = 10:1) to provide a yellow solid (3.5 g, 63% yield). 1H NMR (600 MHz, DMSO-d6, δ): 9.20 (d, J = 18 Hz, 2H), 8.55 (d, J = 18 Hz, 1H), 7.93 (d, J = 12 Hz, 1H), 7.49–7.58 (m, 7H), 7.39 (t, J = 12 Hz, 1H), 6.92–7.05 (m, 3H), 3.56 (s, 3H). 13C NMR (600 MHz, DMSO-d6, δ): 157.15, 150.47, 137.02, 136.31, 132.11, 131.49, 131.00, 130.14, 129.70, 129.40, 129.00, 128.34, 128.21, 127.41, 126.68, 126.62, 125.84, 124.02, 121.92, 121.43, 119.98, 119.33, 119.30, 119.00, 110.92, 54.96. HRMS: 558.9842 [M + H]+ (calcd: 557.98). Anal. Calcd for C28H18Br2N2O: C 60.24, H 3.25, N 5.02; found: C 60.22, H 3.26, N 5.01.
2.3.1 2-(2-Methoxyphenyl)-1,6,9-triphenyl-1H-phenanthro[9,10-d]imidazole (M2)
To a three-necks round-bottom flask, M1 (1.66 g, 3 mmol), phenylboronic acid (2.2 g, 18 mmol), potassium carbonate (10 g, 72 mmol), tetrabutylammonium bromide (0.1 g, 0.3 mmol), toluene (40 mL) and distilled water (25 mL) were added. Then, the mixture was bubbled by nitrogen for 10 min. After that, tetrakis(triphenylphosphine)palladium was added. Under nitrogen atmosphere, the mixture was stirred at 100 °C for 12 h. After cooling to room temperature, the toluene solution was seperated. The aqueous solution was extracted by dichloromethane for three times (50 mL × 3). The organic solvent was removed by rotary evaporation to obtain the crude product. The crude product was purified by column chromatography on silica gel (eluent: dichloromethane/ethyl acetate = 10:1) to provide a yellow solid (1.17 g, 68% yield). 1H NMR (600 MHz, DMSO-d6, δ): 9.30 (t, J = 24 Hz, 2H), 8.73 (d, J = 12 Hz, 1H), 8.10 (d, J = 12 Hz, 1H), 8.03 (d, J = 12 Hz, 2H), 7.94 (d, J = 12 Hz, 2H), 7.70 (d, J = 12 Hz, 1H), 7.49–7.58 (m, 10H), 7.38–7.46 (m, 3H), 7.01 (t, J = 6 Hz, 1H), 6.96 (d, J = 12 Hz, 1H), 6.77 (t, J = 6 Hz, 1H), 3.58 (s, 3H). 13C NMR (600 MHz, DMSO-d6, δ): 157.32, 157.22, 150.03, 140.56, 139.99, 137.47, 137.40, 136.87, 136.55, 132.21, 131.36, 129.51, 129.35, 128.96, 128.91, 128.87, 128.49, 128.05, 127.39, 127.30, 126.96, 126.67, 126.53, 126.24, 125.60, 122.69, 122.47, 121.85, 121.73, 120.85, 120.00, 119.68, 118.77, 115.21, 110.93, 54.96. HRMS: 553.2258 [M + H]+ (calcd: 552.22). Anal. Calcd for C40H28N2O: C 86.93, H 5.11, N 5.07, O 2.89; found: C 86.88, H 5.12, N 5.09, O 2.91.
2.3.2 2-(1,6,9-Triphenyl-1H-phenanthro[9,10-d]imidazol-2-yl)phenol (TPPI)
To a three-necks round-bottom flask, M2 (1.104 g, 2 mmol) and dried dichloromethane (30 mL) were added. Then, boron tribromide (1 mol/L in dichloromethane) (2 mL, 2 mmol) was added. Under nitrogen atmosphere, the mixture was refluxed for 12 h. The mixture changed from light-yellow to purple. After finished, the mixture was cooled to room temperature and methanol was added to quench the residual boron tribromide. Then, sodium bicarbonate solution was added and the mixture was stirred for 10 min. The dichloromethane solution was seperated and the aqueous solution was extracted by dichloromethane for two times (50 mL × 2). Dichloromethane was removed by rotary evaporation to obtain the crude product. The crude product was purified by column chromatography on silica gel (eluent: petroleum/dichloromethane = 1:1) to provide a yellow solid (0.774 g, 72% yield). 1H NMR (600 MHz, CDCl3, δ): 13.80 (s, 1H), 9.02 (s, 1H), 8.96 (s, 1H), 8.80 (d, J = 12 Hz, 1H), 8.03 (d, J = 12 Hz, 1H), 7.67–7.84 (m, 8H), 7.38–7.57 (m, 7H), 7.23 (t, J = 12 Hz, 2H), 7.11–7.16 (m, 2H), 6.78 (d, J = 12 Hz, 1H), 6.53 (t, J = 12 Hz, 1H). 13C NMR (600 MHz, DMSO-d6, δ): 157.37, 149.28, 140.45, 139.86, 137.98, 137.83, 137.18, 131.03, 130.44, 130.40, 130.35, 129.16, 128.98, 128.92, 128.89, 128.80, 128.29, 128.20, 127.52, 127.47, 127.44, 127.34, 126.97, 126.81, 125.79, 125.77, 125.30, 125.25, 125.08, 125.07, 122.61, 122.58, 122.55, 121.98, 121.52, 120.94, 118.30, 116.68, 116.62. HRMS: 539.2114 [M + H]+ (calcd: 538.20). Anal. Calcd for C39H26N2O: C 86.96, H 4.87, N 5.20, O 2.97; found: C 86.97, H 4.86, N 5.22, O 2.95.
2.3.3 Zn(TPPI)2
To a three-necks round-bottom flask, TPPI (1.076 g, 2 mmol) and dried toluene (30 mL) were added. Then, diethylzinc (1 mol/L in toluene) (4 mL, 4 mmol) was added. Under nitrogen atmosphere, the mixture was stirred for 10 h at room temperature. After finished, the solvent and residual diethylzinc were removed by rotary evaporation. The crude product was purified by direct sublimation for twice and a white powder was obtained (0.853 g, 75% yield). 1H NMR (600 MHz, DMSO-d6, δ): 8.47 (s, 2H), 8.11 (d, J = 12 Hz, 2H), 8.04 (s, 2H), 7.87 (d, J = 12 Hz, 4H), 7.70–7.82 (m, 8H), 7.58 (t, J = 6 Hz, 4H), 7.40–7.46 (m, 9H), 7.31 (d, J = 6 Hz, 6H), 7.19 (t, J = 12 Hz, 3H), 7.07 (d, J = 12 Hz, 2H), 6.99 (d, J = 12 Hz, 2H), 6.72 (d, J = 12 Hz, 2H), 6.61 (d, J = 12 Hz, 2H), 6.31 (t, J = 6 Hz, 2H). 13C NMR (600 MHz, DMSO-d6, δ): 167.35, 151.41, 139.91, 139.03, 137.31, 136.99, 136.69, 132.99, 131.80, 130.57, 130.41, 129.99, 129.38, 129.12, 129.07, 129.00, 128.93, 128.54, 128.52, 128.31, 127.54, 127.46, 127.42, 127.35, 127.24, 127.05, 126.81, 126.23, 125.46, 125.30, 122.19, 122.07, 121.33, 121.03, 120.13, 120.04, 119.27, 113.62, 113.14. MALDI-TOF–MS: 1139.539 [M + H]+ (calcd: 1138.32). Anal. Calcd for C78H50N4O2Zn: C 82.13, H 4.42, N 4.91; found: C 82.05, H 4.46, N 4.89.
3 Results and discussion
3.1 Synthesis and thermal properties
Scheme 1 outlines the synthetic procedures of Zn(TPPI)2. The phenanthro[9,10-d]imidazole structure was prepared from phenanthrene-9,10-dione. Then, benzene rings were added through Suzuki coupling reaction. The demethylation was carried out by boron tribromide to provide the ligand. Sequentially, the ligand reacted with diethylzinc to obtain crude product. Zn(TPPI)2 was further purified by vacuum sublimation and a white powder was obtained. Finally, it was fully characterized by 1H NMR, 13C NMR, mass spectra and element analysis (Fig. S1-S12). Every reaction could be carried out easily and had high yield. This was favorable for its practical application.
Thermal properties of Zn(TPPI)2 were investigated using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. S13 and Table 1). Zn(TPPI)2 exhibited high thermal stability. From TGA curve, its decomposition temperature (Td) (corresponding to 5% weight loss) was determined to be 495 °C. No glass transition was observed at its DSC curve. Good thermal stability favors thermal evaporation preparation of uniform films which can increase the device efficiency. The atomic force microscope (AFM) image of Zn(TPPI)2 film exhibits low root-mean-square (RMS) roughness (0.509 nm), indicating that this film is smooth (Fig. S14). Meanwhile, good thermal stability can also prolong the device's operational lifetime.
3.2 Electrochemical and charge-transporting properties
The electrochemical properties of Zn(TPPI)2 were investigated by the cyclic voltammetry (CV) measurement in N,N-dimethylformamide (DMF) (Fig. 1a). Zn(TPPI)2 exhibited irreversible oxidation behavior. From the oxidation peak, its highest occupied molecular orbital (HOMO) energy level was determined to be − 5.33 eV according to the equation of (EHOMO/LUMO = − (E[onset, ox/red vs Fc+/Fc] + 4.8) eV. This HOMO energy level was close to those of common hole-transporting materials, indicating that hole could be easily injected from hole-transporting layers to Zn(TPPI)2 layer. No reduction peak was detected. Its lowest unoccupied molecular orbital (LUMO) energy level was determined to be − 2.15 eV from its HOMO and absorption spectra.

a Cyclic voltammogram of Zn(TPPI)2 in DMF (scan rate: 100 mV s−1; electrolyte: tetrabutylammonium hexafluorophosphate, 0.1 M; test method: three-electrode method). b Current density versus voltage characteristics of hole-only and electron-only devices of Zn(TPPI)2
To investigate the charge-transporting ability of Zn(TPPI)2, the hole-only and electron-only devices with the structures of [ITO/Zn(TPPI)2 (60 nm)/TAPC (20 nm)/Al] for hole-only and [ITO/TmPyPB (20 nm)/Zn(TPPI)2 (60 nm)/LiF (1.5 nm)/Al] for electron-only were fabricated. The device structures were shown in Fig. S15. Results were shown in Fig. 1b. Compared with the aluminum cathode (LUMO: − 4.3 eV), the much higher LUMO (− 2.0 eV) of TAPC brought about large electron-injection barrier. Hence, only holes could be injected to obtain pure hole current. Similarly, the hole-blocking material, TmPyPB (HOMO: − 6.7 eV), could prevent the hole-injection from ITO (HOMO: − 4.8 eV) to endow the device with pure electron current. From Fig. 1b, based on the space charge limited current (SCLC) method, the hole and electron mobilities of Zn(TPPI)2 were calculated to be 2.46 × 10–11 cm2 V−1 s−1 and 2.61 × 10–11 cm2 V−1 s−1 respectively. Although these two mobilities were comparable, they were obtained at low voltage (< 1 V) which was far from the device's working voltage (> 2.5 V). At the device's working voltage, the hole current was much higher than the electron current, indicating that Zn(TPPI)2 was a hole-type material.
Meanwhile, the hole-only and electron-only devices of doped films (Zn(TPPI)2: 3wt% Ir(bt)2(acac)) (Zn(TPPI)2: 10% Ir(piq)3) with the structures of [ITO/doped films (60 nm)/TAPC (20 nm)/Al] for hole-only and [ITO/TmPyPB (20 nm)/doped films (60 nm)/LiF (1.5 nm)/Al] for electron-only were also fabricated. Results were shown in Fig. S16. After Ir(bt)2(acac) and Ir(piq)3 were introduced, the current densities of emitting layers increased. Based on the space charge limited current (SCLC) method, the hole and electron mobilities of Ir(bt)2(acac)-doped film were calculated to be 7.85 × 10–11 cm2 V−1 s−1 and 1.76 × 10–10 cm2 V−1 s−1 respectively; the hole and electron mobilities of Ir(piq)3-doped film were calculated to be 1.61 × 10–9 cm2 V−1 s−1 and 2.28 × 10–8 cm2 V−1 s−1 respectively.
3.3 Photophysical properties
The room-temperature UV–Vis absorption and photoluminescence (PL) spectra in dichloromethane (CH2Cl2) solution (10–5 M) and in neat film (100 nm) as well as the low-temperature PL spectra in CH2Cl2 solution (10–5 M) and phosphorescence spectra in 2-methyltetrahydrofuran (2-MeTHF) solution (10–5 M) at 77 K of Zn(TPPI)2 were shown in Fig. 2. According to previous work [27], the absorption peaks below 300 nm, around 340 nm and around 375 nm should be ascribed to the benzene rings, the π…π* transition of phenanthro[9,10-d]imidazole, and the delocalized π…π* transition from the 2-substituted phenolate to phenanthro[9,10-d]imidazole ring respectively. The molar extinction coefficient in dichloromethane solution was 2.67 × 105 L/(mol·cm). From the absorption spectra, the energy gap between S1,0 and S0,0 was estimated to be 3.18 eV.

Room-temperature UV–Vis absorption and photoluminescence (PL) spectra in CH2Cl2 solution (10−5 M) and in neat film (100 nm) as well as the low-temperature PL spectra in CH2Cl2 solution (10−5 M) and phosphorescence spectra in 2-methyltetrahydrofuran (2-MeTHF) solution (10−5 M) at 77 K of Zn(TPPI)2
To investigate the ground-state and excited-state characteristics of Zn(TPPI)2, its absorption and PL spectra in different solvents (hexane, toluene, 1,4-dioxane, triethylamine, butylether, isopropylether, diethyl ether, ethyl acetate, tetrahydrofuran, dichloromethane, acetone) were measured (Fig. 3). With the increase of solvent polarity, the absorption and PL spectra were almost the same and no obvious red-shift was observed, indicating that the ground-state dipole moment of Zn(TPPI)2 was small and its excited-state was not from charge transfer. In different solvents, all the PL spectra exhibited fine vibrational structure, illustrating that its singlet emission was from locally-excited state. Similarly, its phosphorescence spectra also showed fine structure. Hence, its triplet emission was also from locally-excited state. The transient photoluminescence (PL) decay characteristic of Zn(TPPI)2 film was measured (Fig. S17). From this curve, the photoluminescence lifetime of Zn(TPPI)2 was determined to be 2.96 ns.

Room-temperature a UV–Vis absorption and b photoluminescence (PL) spectra of Zn(TPPI)2 in different solvents (10–5 M) (hexane, toluene, 1,4-dioxane, triethylamine, butylether, isopropylether, diethyl ether, ethyl acetate, tetrahydrofuran, dichloromethane, acetone)
Zn(TPPI)2 exhibited blue emission in both solution (467, 473 nm) and film state (447 nm) (Fig. 2). Compared with the film emission, the solution emission showed a redshift. For most of fluorescent materials, with the increase of aggregation degree, the delocalization degree would increase and the emission peak would have a red shift. However, if the molecule had some composition groups which could rotate and vibrate freely, the variation tendency might be different. For such molecule, the rotation and vibration of compostion groups consumed much energy. When the molecule aggregated, these unfavorable motions would be limited and the wasted energy could be used for emission. This principle was popular in aggregation-induced emission (AIE) materials. Delocalization and wasted energy reduction competed. If the influence of wasted energy reduction was greater than that of delocalization, the emission peak would have a blue shift. In contrast, the emission peak would have a red shift. To investigate the delocalization influence of Zn(TPPI)2, photoluminescence (PL) spectra (10−4, 10−5, 10−6 M) of Zn(TPPI)2 in dichloromethane solution were measured. Results were shown in Fig. S18. With the increase of concentration, the PL intensity increased but the PL peak almost had no change, indicating that with the increase of aggregation degree, the delocalization influence was not obvious. This was because benzene rings in Zn(TPPI)2 efficiently prevented the close packing of molecules. For wasted energy reduction, from the molecular structure of Zn(TPPI)2, it could be seen that Zn(TPPI)2 contained six benzene rings which could rotate and vibrate freely. Much energy was consumed by these molecular motions. In film, the aggregation state limited these unfavorable motions and the wasted energy could be used for emission. Delocalization influence was weak and wasted energy reduction could exceed it. Hence, compared with the solution emission, the film emission showed a blue-shift.
Similarly, compared with the solution emission at room temperature, the emission (452, 463 nm) at 77 K also exhibited a blue-shift because those unfavorable motions stopped at 77 K. From the onsets of fluorescence and phosphorescence spectra at 77 K, the lowest singlet excited state (S1) and lowest triplet excited state (T1) energy levels of Zn(TPPI)2 were calculated to be 2.90 eV and 2.59 eV, respectively. Hence, its singlet–triplet splitting (∆EST) was 0.31 eV. Finally, the photoluminescence quantum yields (PLQYs) of Zn(TPPI)2 in dichloromethane (10–5 mol/L) and pure film were 68% and 21%, respectively.
3.4 Electroluminescent properties
To investigate the luminescent property of Zn(TPPI)2, undoped device with the structure of [ITO/HATCN (10 nm)/TAPC (30 nm)/TCTA (4 nm)/Zn(TPPI)2 (15 nm)/TmPyPB (25 nm)/LiF (1 nm)/Al] (Device B1) was fabricated. In this device, indium tin oxide (ITO), dipyrazino [2,3-f:2′,3′-h] quinoxaline-2,3,6,7,10,11-hexacarbonitrile (HATCN), 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC), 4,4′,4″-tris (carbazol-9-yl)-triphenylamine (TCTA), 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB), lithium fluoride (LiF) and aluminium (Al) were utilized as the anode, hole-injection layer, hole-transporting layer, hole-transporting layer, electron-transporting layer, electron-injection layer and cathode, respectively. Undoped Zn(TPPI)2 layer was employed as the emitting layer. The energy level diagram and chemical structures of the materials used in OLEDs were shown in Fig. 4. The current density–voltage-luminance (J-V-L) characteristics, efficiencies versus luminance curves, EL spectra as well as external quantum efficiencies (EQE) were shown in Fig. 5 and Table 2. Device B1 displayed blue emission (CIE: 0.17, 0.18). Its maximum brightness (Lmax), maximum current efficiency (ηc.max), maximum power efficiency (ηp.max) and maximum external quantum efficiency (ηext.max) were 1153 cd/m2, 3.79 cd/A, 3.97 lm/W and 2.47% respectively. For zinc-complex blue emitters, this performance is high [23,24,25]. Meanwhile, with the variation of driving voltage, device B1 exhibited good electroluminescent color stability which was important for practical application (Fig. S19a, b).

Energy level diagram and chemical structures of the materials used in OLEDs

a Current density–voltage–luminance (J–V–L) curves of B1, Y1 and R1. b Current efficiency–luminance–power efficiency (CE–L–PE) curves of B1, Y1 and R1. c External quantum efficiency–luminance curves of B1, Y1 and R1. d EL spectra of B1, Y1 and R1 at the luminance of 100 cd/m2
Results of single-carrier devices and photophysical measurement indicated that Zn(TPPI)2 possessed the potential to be an excellent host. To investigate the host property of Zn(TPPI)2, phosphorescent devices with the structures of [ITO/HATCN (10 nm)/TAPC (30 nm)/TCTA (4 nm)/Zn(TPPI)2: 3wt% Ir(bt)2(acac) or 10% Ir(piq)3 (15 nm)/TmPyPB (25 nm)/LiF (1 nm)/Al] (Device Y1 for Ir(bt)2(acac) and R1 for Ir(piq)3) were fabricated. Bis(2-phenyl-benzothiazole-C2, N)(acetylacetonate)iridium(III) (Ir(bt)2(acac)) and tris(1-phenylisoquinoline)iridium(III) (Ir(piq)3) were utilized as yellow–red and red phosphorescent emitters respectively. The molecular structures in devices and their energy levels were shown in Fig. 4. The current density–voltage–luminance (J–V–L) characteristics, efficiencies versus luminance curves, EL spectra as well as external quantum efficiencies (EQE) were shown in Fig. 5 and Table 2.
As shown in Fig. 5d, device Y1 showed yellow–red emission and no emission peak of Zn(TPPI)2 was observed, indicating that the energy transfer from Zn(TPPI)2 to Ir(bt)2(acac) was complete. With the increase of driving voltage, the electroluminescent (EL) spectra exhibited good stability (Fig. S19c, d). Device Y1 showed low turn-on voltage (Von) (2.6 V). This should be attributed to the appropriate energy levels of Zn(TPPI)2 and Ir(bt)2(acac). First, the HOMO (− 5.33 eV) energy level of Zn(TPPI)2 was favorable for hole injection from hole-transporting layer. Second, the LUMO (− 2.9 eV) energy level of Ir(bt)2(acac) endowed it with good carrier trap ability and electron could be injected into phosphorescent dopant directly and easily from electron-transporting layer. The Lmax, ηc.max, ηp.max and ηext.max were 12,526 cd/m2, 58.35 cd/A, 70.52 lm/W and 20.82% respectively. The high device efficiencies should be ascribed to the following reasons. First, as discussed above, the appropriate energy levels of Zn(TPPI)2 and Ir(bt)2(acac) were favorable for hole and electron injection from hole- and electron-transporting layers. Hence, charge could enter the emitting layer easily. Second, good thermal stability favored thermal evaporation preparation of uniform films which could increase the device efficiency. Table S1 compared this work and previous publications for Ir(bt)2(acac)-doped devices [28,29,30,31,32,33,34,35,36]. It could be seen that the highest efficiencies of Zn(TPPI)2-based device (Y1) were comparable to the best results of previous publications. However, its roll-off of efficiency was much bigger, indicating that Zn(TPPI)2 might not match with Ir(bt)2(acac).
To further compare Zn(TPPI)2 with common host, phosphorescent device with the structure of [ITO/HATCN (10 nm)/TAPC (30 nm)/TCTA (4 nm)/mCP: 3wt% Ir(bt)2(acac) (15 nm)/TmPyPB (25 nm)/LiF (1 nm)/Al] (Device D1) was fabricated. Common host, N, N-dicarbazolyl-3,5-benzene (mCP), was utilized for comparison. The current density–voltage–luminance (J–V–L) characteristics, efficiencies versus luminance curves, EL spectra as well as external quantum efficiencies (EQE) were shown in Fig. S20 and Table 2. The Von, Lmax, ηc.max, ηp.max and ηext.max of device D1 were 3.2 V, 28,100 cd/m2, 50.42 cd/A, 44.18 lm/W and 17.84% respectively. Obviously, the performance of device D1 was lower than that of device Y1, indicating that Zn(TPPI)2 was superior to mCP as host in Ir(bt)2(acac)-based device.
Except for yellow–red phosphorescent device, Zn(TPPI)2 also exhibited high performance in red phosphorescent device (Fig. 5). As shown in Fig. 5a–c and Table 2, the Von, Lmax, ηc.max, ηp.max and ηext.max of device R1 were 2.8 V, 11,000 cd/m2, 15.89 cd/A, 16.09 lm/W and 19.05% respectively. Meanwhile, device R1 exhibited low roll-off of efficiencies. The high device efficiencies and low roll-off of efficiencies should be ascribed to the same above reasons in device Y1. Table 3 compared this work and previous publications for Ir(piq)3-doped devices [19, 37,38,39,40,41,42,43,44,45,46]. It could be seen that for Ir(piq)3-doped device, Zn(TPPI)2-based device (R1) not only exhibited performance comparable to the best results of previous publications, but also displayed much smaller roll-off of efficiency, indicating that Zn(TPPI)2 was a good host.
4 Conclusion
In summary, we reported a novel phenanthro[9,10-d]imidazole-zinc coordination complex. This compound exhibited series of fascinating characteristics: excellent thermal stability (Td: 495 °C), appropriate HOMO (− 5.33 eV) energy level for efficient hole injection from transporting layer, small singlet–triplet splitting (0.31 eV). These characteristics met the requirements of an excellent host. Yellow–red (58.35 cd/A, 70.52 lm/W, 20.82%) and red (15.89 cd/A, 16.09 lm/W and 19.05%) phosphorescent devices which utilized Zn(TPPI)2 as host were fabricated. The performance of red device was comparable to the best results of previous publications on corresponding emitter. Meanwhile, the devices displayed good electroluminescent color stability. These results demonstrated that Zn(TPPI)2 was a good host material. Finally, Zn(TPPI)2 was also a good blue emitter and its undoped blue device displayed high efficiencies (3.79 cd/A, 3.97 lm/W, 2.47%) in zinc-complex blue emitters.
References
H. Na, T.S. Teets, Highly luminescent cyclometalated iridium complexes generated by nucleophilic addition to coordinated isocyanides. J. Am. Chem. Soc. 140(20), 6353–6360 (2018)
T. Fleetham, Y.L. Ji, L. Huang, T.S. Fleetham, J. Li, Efficient and stable single-doped white OLEDs using a palladium-based phosphorescent excimer. Chem. Sci. 8(12), 7983–7990 (2017)
N. Su, G.Z. Lu, Y.X. Zheng, Highly efficient green electroluminescence of iridium(III) complexes based on (1H-pyrazol-5-yl)pyridine derivatives ancillary ligands with low efficiency roll-off. J. Mater. Chem. C 6(21), 5778–5784 (2018)
X.L. Yang, B. Jiao, J.S. Dang, Y.H. Sun, Y. Wu, G.J. Zhou, W.Y. Wong, Achieving high-performance solution-processed orange OLEDs with the phosphorescent cyclometalated trinuclear Pt(II) complex. ACS Appl. Mater. Interfaces 10(12), 10227–10235 (2018)
J. Zhao, Z. Feng, D. Zhong, X. Yang, Y. Wu, G. Zhou, Z. Wu, Cyclometalated platinum complexes with aggregation-induced phosphorescence emission behavior and highly efficient electroluminescent ability. Chem. Mater. 30(3), 929–946 (2018)
Y.Q. Miao, P. Tao, L. Gao, X.L. Li, L.W. Wei, S.J. Liu, H. Wang, B.S. Xu, Q. Zhao, Highly efficient chlorine functionalized blue iridium(III) phosphors for blue and white phosphorescent organic light-emitting diodes with the external quantum efficiency exceeding 20%. J. Mater. Chem. C 6(25), 6656–6665 (2018)
Z.W. Liu, J. Qiu, F. Wei, J.Q. Wang, X.C. Liu, M.G. Helander, S. Rodney, Z.B. Wang, Z.Q. Bian, Z.H. Lu, M.E. Thompson, C.H. Huang, Simple and high efficiency phosphorescence organic light-emitting diodes with codeposited copper(I) emitter. Chem. Mater. 26, 2368–2373 (2014)
K.Y. Hwang, M.H. Lee, H. Jang, Y. Sung, J.S. Lee, S.H. Kim, Y. Do, Aluminium-salen luminophores as new hole-blocking materials for phosphorescent OLEDs. Dalton Trans. 14(14), 1818–1820 (2008)
P. Kathirgamanathan, S. Surendrakumar, J. Antipan-Lara, S. Ravichandran, Y.F. Chan, V. Arkley, S. Ganeshamurugan, M. Kumaraverl, G. Paramswara, A. Partheepan, V.R. Reddy, D. Bailey, A.J. Blake, Novel lithium Schiff-base cluster complexes as electron injectors: synthesis, crystal structure, thin film characterisation and their performance in OLEDs. J. Mater. Chem. 22(13), 6104–6116 (2012)
A.S. Burlov, V.G. Vlasenko, A.V. Dmitriev, V.V. Chesnokov, A.I. Uraev, D.A. Garnovskii, Y.V. Zubavichus, A.L. Trigub, I.S. Vasilchenko, D.A. Lypenko, E.I. Mal’tsev, T.V. Lifintseva, G.S. Borodkin, Synthesis, structure, photo- and electroluminescent properties of zinc(II) complexes with aminomethylene derivatives of 1-phenyl-3-methyl-4-formylpyrazol-5-one and 3-and 6-aminoquinolines. Synthetic Met. 203, 156–163 (2015)
Y.G. Li, X. Gao, L. Wang, G.L. Tu, Deep-red organic light-emitting diodes with stable electroluminescent spectra based on zinc complex host material. RSC Adv. 7(64), 40533–40538 (2017)
M.E. Burin, V.A. Ilichev, A.P. Pushkarev, D.L. Vorozhtsov, S.Y. Ketkov, G.K. Fukin, M.A. Lopatin, A.A. Nekrasov, M.N. Bochkarev, Synthesis and luminescence properties of lithium, zinc and scandium 1-(2-pyridyl)naphtholates. Org. Electron. 13(12), 3203–3210 (2012)
G. Cheng, G.K.M. So, W.P. To, Y. Chen, C.C. Kwok, C.S. Ma, X.G. Guan, X. Chang, W.M. Kwok, C.M. Che, Luminescent zinc(II) and copper(I) complexes for high-performance solution-processed monochromic and white organic light-emitting devices. Chem. Sci. 6(8), 4623–4635 (2015)
Q.C. Zhang, H. Xiao, X. Zhang, L.J. Xu, Z.N. Chen, Luminescent oligonuclear metal complexes and the use in organic light-emitting diodes. Coordin. Chem. Rev. 378, 121–133 (2019)
J. Zhao, F.F. Dang, B. Liu, Y. Wu, X.L. Yang, G.J. Zhou, Z.X. Wu, W.Y. Wong, Bis-Zn-II salphen complexes bearing pyridyl functionalized ligands for efficient organic light-emitting diodes (OLEDs). Dalton Trans. 46(18), 6098–6110 (2017)
F. Dumur, L. Beouch, M.A. Tehfe, E. Contal, M. Lepeltier, G. Wantz, B. Graff, F. Goubard, C.R. Mayer, J. Lalevee, D. Gigmes, Low-cost zinc complexes for white organic light-emitting devices. Thin Solid Films 564, 351–360 (2014)
M.G. Kaplunov, S.S. Krasnikova, S.L. Nikitenko, N.L. Sermakasheva, I.K. Yakushchenko, Exciplex electroluminescence and photoluminescence spectra of the new organic materials based on zinc complexes of sulphanylamino-substituted ligands. Nanoscale Res. Lett. 7, 206 (2012)
Y. Sakai, Y. Sagara, H. Nomura, N. Nakamura, Y. Suzuki, H. Miyazaki, C. Adachi, Zinc complexes exhibiting highly efficient thermally activated delayed fluorescence and their application to organic light-emitting diodes. Chem. Commun. 51(15), 3181–3184 (2015)
R.J. Wang, L.J. Deng, M. Fu, J.L. Cheng, J.Y. Li, Novel Zn-II complexes of 2-(2-hydroxyphenyl)benzothiazoles ligands: electroluminescence and application as host materials for phosphorescent organic light-emitting diodes. J. Mater. Chem. 22(44), 23454–23460 (2012)
M. Janghouri, S. Minasian, M. Mahmoudi, S. Eyvazi, Photo/electroluminescence and electron transport properties of new zinc complexes. Opt. Mater. 89, 488–493 (2019)
Y.P. Huo, J.G. Lu, T.H. Lu, X.M. Fang, X.H. Ouyang, L. Zhang, G.Z. Yuan, Comparative studies on OLED performances of chloro and fluor substituted Zn(II) 8-hydroxyquinolinates. New J. Chem. 39(1), 333–341 (2015)
R. Lakshmanan, N.C. Shivaprakash, S. Sindhu, Switching from sky blue to deep green fluorescent Zn(II) complexes for OLEDs applications. J. Lumin. 196, 136–145 (2018)
F. Dumur, Zinc complexes in OLEDs: an overview. Synthetic Met. 195, 241–251 (2014)
X.J. Xu, Y. Liao, G. Yu, H. You, C.G. Di, Z.M. Su, D.G. Ma, Q. Wang, S.Y. Li, S.Q. Wang, J.P. Ye, Y.Q. Liu, Charge carrier transporting, photoluminescent, and electroluminescent properties of zinc(II)-2-(2-hydroxyphenyl)benzothiazolate complex. Chem. Mater. 19(7), 1740–1748 (2007)
S.G. Roh, Y.H. Kim, K.D. Seo, D.H. Lee, H.K. Kim, Y.I. Park, J.W. Park, J.H. Lee, Synthesis, photophysical, and electroluminescent device properties of Zn(II)-chelated complexes based on functionalized benzothiazole derivatives. Adv. Funct. Mater. 19(10), 1663–1671 (2009)
C.S. Oh, J.Y. Lee, High triplet energy Zn complexes as host materials for green and blue phosphorescent organic light-emitting diodes. Dyes Pigments 99(2), 374–377 (2013)
K. Wang, F.C. Zhao, C.G. Wang, S.Y. Chen, D. Chen, H.Y. Zhang, Y. Liu, D.G. Ma, Y. Wang, High-performance red, green, and blue electroluminescent devices based on blue emitters with small singlet-triplet splitting and ambipolar transport property. Adv. Funct. Mater. 23(21), 2672–2680 (2013)
K. Gao, K.K. Liu, X.L. Li, X.Y. Cai, D.J. Chen, Z.D. Xu, Z.Z. He, B.B. Li, Z.Y. Qiao, D.C. Chen, Y. Cao, S.J. Su, An ideal universal host for highly efficient full-color, white phosphorescent and TADF OLEDs with a simple and unified structure. J. Mater. Chem. C 5(39), 10406–10416 (2017)
T. Chatterjee, W.Y. Hung, W.F. Tang, H.F. Chen, K.T. Wong, Carbazole-bridged triphenylamine-bipyridine bipolar hosts for high-efficiency low roll-off multi-color PhOLEDs. Org. Electron. 50, 204–212 (2017)
Y.Q. Miao, K.X. Wang, L. Gao, B. Zhao, H. Wang, F.R. Zhu, B.S. Xu, D.G. Ma, Precise manipulation of the carrier recombination zone: a universal novel device structure for highly efficient monochrome and white phosphorescent organic light-emitting diodes with extremely small efficiency roll-off. J. Mater. Chem. C 6(30), 8122–8134 (2018)
T.L. Chiu, H.J. Chen, T.C. Lin, H.J. Gau, Y.H. Hsieh, J.J. Huang, L.K. Yun, J.H. Lee, M.K. Leung, Colour stability of blue-green and white phosphorescent organic light-emitting diode employing a 9-(2-(4,5-diphenyl-4H-1,2,4-triazol-3-yl)phenyl)-9H-carbazole host. Dyes Pigments 141, 463–469 (2017)
C.J. Zheng, C.L. Liu, K. Wang, S.L. Tao, H. Lin, C.S. Lee, Novel bipolar host for highly efficient green, yellow, orange, red and deep-red phosphorescent organic light-emitting devices. Sci. China Chem. 60(4), 504–509 (2017)
Y.X. Hu, X. Xia, W.Z. He, H.J. Chi, Y. Dong, G.Y. Xiao, Y.L. Lv, X. Li, D.Y. Zhang, Novel Ir(III) complexes ligated with 2-(2,6-difluoropyridin-3-yl)benzo d thiazole for highly efficient OLEDs with mild efficiency roll-off. Dyes Pigments 166, 254–259 (2019)
D. Liu, R.J. Yao, R.Z. Dong, F.J. Jia, M. Fu, A strategy to increase phosphorescent efficiency without perturbing emission color for benzothiazole-containing iridium phosphors. Dyes Pigments 145, 528–537 (2017)
S.H. Ye, L. Li, M. Zhang, Z. Zhou, M.H. Quan, L.F. Guo, Y. Wang, M. Yang, W.Y. Lai, W. Huang, Pyridine linked fluorene hybrid bipolar host for blue, green, and orange phosphorescent organic light-emitting diodes toward solution processing. J. Mater. Chem. C 5(45), 11937–11946 (2017)
J.H. Jang, H.J. Park, J.Y. Park, H.U. Kim, D.H. Hwang, Orange phosphorescent Ir(III) complexes consisting of substituted 2-phenylbenzothiazole for solution-processed organic light-emitting diodes. Org. Electron. 60, 31–37 (2018)
H.H. Chou, C.H. Cheng, A highly efficient universal bipolar host for blue, green, and red phosphorescent OLEDs. Adv. Mater. 22(22), 2468–2471 (2010)
S.J. Su, C. Cai, J. Takamatsu, J. Kido, A host material with a small singlet-triplet exchange energy for phosphorescent organic light-emitting diodes: guest, host, and exciplex emission. Org. Electron. 13(10), 1937–1947 (2012)
S.J. Su, C. Cai, J. Kido, RGB phosphorescent organic light-emitting diodes by using host materials with heterocyclic cores: Effect of nitrogen atom orientations. Chem. Mater. 23(2), 274–284 (2011)
S.J. Su, C. Cai, J. Kido, Three-carbazole-armed host materials with various cores for RGB phosphorescent organic light-emitting diodes. J. Mater. Chem. 22(8), 3447–3456 (2012)
Y.Y. Lyu, J. Kwak, W.S. Jeon, Y. Byun, H.S. Lee, D. Kim, C. Lee, K. Char, Highly efficient red phosphorescent OLEDs based on non-conjugated silicon-cored spirobifluorene derivative doped with Ir-complexes. Adv. Funct. Mater. 19(3), 420–427 (2009)
H. Kim, Y. Byun, R.R. Das, B.K. Choi, P.S. Ahn, Small molecule based and solution processed highly efficient red electrophosphorescent organic light emitting devices. Appl. Phys. Lett. 91(9), 093512 (2007)
R.D. Guo, W.Z. Zhang, Q. Zhang, X.L. Lv, L. Wang, Efficient deep red phosphorescent OLEDs using 1,2,4-thiadiazole core-based novel bipolar host with low efficiency roll-off. Front. Optoelectron. 11(4), 375–384 (2018)
X. Wang, J. Zhou, J. Zhao, Z.Y. Lu, J.S. Yu, High performance fluorescent and phosphorescent organic light-emitting diodes based on a charge-transfer-featured host material. Org. Electron. 21, 78–85 (2015)
S. Grigalevicius, D. Tavgeniene, G. Krucaite, R. Griniene, W.C. Li, D. Luo, C.H. Chang, 9,9’-Bis(2,2-diphenylvinyl)-3,3’-bicarbazole as low cost efficient hole transporting material for application in red PhOLEDs. Dyes Pigments 152, 100–104 (2018)
C.H. Chang, R. Griniene, Y.D. Su, C.C. Yeh, H.C. Kao, J.V. Grazulevicius, D. Volyniuk, S. Grigalevicius, Efficient red phosphorescent OLEDs employing carbazole-based materials as the emitting host. Dyes Pigments 122, 257–263 (2015)
Acknowledgements
This work was supported by National Key Research and Development Project of China (2018YFB0407102), National Natural Science Foundation of China (51903026, 61505018, U1663229, 21603020, 61705026, 51903027), Financial Projects of Sichuan Science and Technology Department (2018ZYZF0062), Natural Science Foundation of Chongqing Science & Technology Commission (cstc2020jcyj-msxmX0474, cstc2017zdcy-yszxX0004, cstc2019jcyjjq0092, cstc2018jszx-cyzdX0137, cstc2016jcyjA0577, cstc2018jcyjAX0212, cstc2019jcyjjqx0021, cstc2018jcyjAX0292), Science and Technology Innovation Leader Plan of Chongqing (T04040012), Science and Technology Research Program of Chongqing Municipal Education Commission (KJQN201901314, KJZD-K201901302, KJ1711269, KJQN201801332, KJQN202001304, KJQN201801317), the Chongqing University Outstanding Achievement Transformation Projects (KJZH17130), Overseas Returnees Support Program for Innovation and Entrepreneurship of Chongqing (cx2018136), Public Service Platform for Industrialization of Technological Innovation Achievements in the Field of Robot and Intelligent Manufacturing in Chongqing (2019-00900-1-1). This work is also sponsored by Sichuan Province Key Laboratory of Display Science and Technology.
Author information
Authors and Affiliations
Contributions
The experimental work was done by the first author. Data verification and guidance were done by other authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, H., Yu, J., Chen, S. et al. Novel phenanthro[9,10-d]imidazole-zinc complex as a host for high-performance OLEDs. J Mater Sci: Mater Electron 32, 22459–22471 (2021). https://doi.org/10.1007/s10854-021-06732-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06732-6