
Abstract
The prompt recombination between photogenerated electrons and holes is the common problem for improving the hydrogen evolution performance of a photocatalyst, which could be solved greatly by composite co-catalysis. Herein, a simple hydrothermal reaction was utilized to prepare g-C3N4/Ni9S8 composite photocatalysts. Through electroless nickel plating, Ni9S8 nanostructure was homogeneously grown onto the g-C3N4 surface by using sodium hypophosphite as reducing agent. With the optimum loading amount of Ni9S8, the acquired composite, compared with the raw g-C3N4, presented a significant increase in hydrogen evolution rate under visible light irradiation, which was measured as 355.7 μmol g−1 h−1 at 7 °C, being 21.2 times that of raw g-C3N4. The mechanism for the hydrogen evolution reaction over the present g-C3N4/Ni9S8 composite photocatalysts was discussed in detail.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, human beings are suffering from energy shortage, environmental pollution and extreme climate, because the increasing large-scale use of fossil energy will cause the depletion of easily available energy sources, giving out a pessimistic perspective for the reserve of fossil energy, and release a lot of waste gases and greenhouse gases, resulting in great impact on the earth’s ecology [1]. Therefore, the development of new energy is very urgent for mankind. Hydrogen energy, as a pearl of new energy, has attracted much attention because of its high calorific value, no carbon emission and facile recyclability [2]. And since 1972 when Fujishima and Honda discovered that TiO2 could decompose water into hydrogen and oxygen under ultraviolet light [3], it seems for human beings to find a shortcut to solve the energy problem once and for all. Thus now photocatalysis is such an exciting technology that attracts numerous researchers to study.
It is soon recognized by scientists that photocatalysis is based on the photoelectric conversion of semiconductors, and ultimately the conversion of light into chemical energy [4]. Therefore, the exploration of semiconductor photocatalysts has been rapidly launched. Today, many photocatalytic semiconductors such as ZnO, Ga2O3, SrTiO3 and ZnS [5,6,7,8,9] have been found to be similar with TiO2 in photocatalytic decomposition of water. However, these semiconductor photocatalysts can only respond to ultraviolet light because of their wide bandgap. To effectively make use of sunlight (containing only 5% ultraviolet light), which is the inexhaustible energy source for human beings, researchers have to search for narrow bandgap semiconductors for photocatalytic hydrogen evolution, and have developed some, such as CdS, MoS, Cu2O and many other [10,11,12]. But their poor photochemical stability, heavy metal pollution on water and/or less earth reserves limit their large-scale application [13].
In 2009, Wang et al. first found that g-C3N4 has the ability of photocatalytic decomposition of water under visible light [14]. Such ability of this kind of organic semiconductors has soon aroused the great interest of scientists. In literature, a large number of studies have shown that g-C3N4 owns a bandgap of roughly 2.78 eV, which can respond to light sources with wavelength below 460 nm [15]. At the same time, it has the advantages of cadenced carbon-to-nitrogen framework (thus providing more active reaction sites for electron donor/acceptors), strong photochemical stability, low-cost, easy availability, non-toxicity and pollution free for environment [16,17,18]. In spite of these virtues, however, there are still some disadvantages on such organic semiconductors for photocatalytical hydrogen evolution, such as high carrier recombination rate due to a large number of intrinsic defects like N and C vacancies, poor electrical conductivity, limited ability to use light source with wavelength only below 460 nm, as well as difficultly dispersible layered structure, which seriously degrades the photocatalytic activity [13].
To reduce the recombination of photogenerated carriers (holes and electrons) in photocatalysts, co-catalysis is an excellent solution [19]. For example, noble metals like Au, Pt, Pd, Rh and so on have excellent catalytic effect due to their unique surface plasmon effect, which can also serve as good co-catalysts [20]. Regrettably, noble metals cannot widely be used in photocatalytic industry due to their high cost and scarce reserves. Therefore, it is very important to find low-cost co-catalysts for photocatalytic hydrogen evolution. In addition, by combining different semiconductor photocatalysts together to form a built-in electric field, photogenerated electrons are easily transferred from the optical semiconductor to the co-catalytic semiconductor. At this time, because of the Schottky barrier, the photogenerated electrons are difficult to return to the original semiconductor catalyst, thus the separation of photogenerated carriers is realized [15]. Meanwhile, for the co-catalytic semiconductor, low overpotential and good conductivity are very helpful for hydrogen evolution [21]. Therefore, it is a feasible idea to find a semiconductor with low overpotential and good conductivity to replace noble metals.
In literature, NiS has been regarded as an excellent co-catalyst in photocatalytic hydrogen evolution, and hopefully it can replace noble metals. This is due to the following advantages of NiS: (1) high-conductivity due to low bandgap, (2) low surface work function that can effectively reduce the reaction activation energy or over potential, (3) high power conversion efficiency, and (4) easy preparation, low-cost as well as environmental friendliness [10]. Therefore, a lot of researches have been executed on the co-catalysis of NiS in photocatalysis, such as CdS/NiS, TiO2/NiS, g-C3N4/NiS and MoO3/NiS [22,23,24,25]. However, since nickel sulfides can exist in different component including NiS, NiS2, Ni3S4, Ni4S3, Ni9S8 and so forth [26], it is difficult for researchers to study them fully. Among them, Ni9S8 is less involved because it is not easily synthesized. As far as we know, no one has reported any composite of Ni9S8 and g-C3N4. Moreover, Ni9S8 has a near zero bandgap compared with NiS, which implies that Ni9S8 has better conductivity and is more conducive to the transmission of photogenerated electrons, and thus the composite of Ni9S8 can effectively improve the photocatalytic activity of MoS2 [27]. In addition, Yang et al. reported that Ni9S8 has a low overpotential and can easily expose surface active sites [28]. Therefore, it is much desirable to prepare the composite of g-C3N4 and Ni9S8 on photocatalytic hydrogen evolution.
For the synthesis of the composites of g-C3N4 with nickel sulfides, several methods including hydrothermal synthesis, calcination, precipitation and photodeposition have been proposed [29,30,31,32]. Among them, the calcination methods are energy-consuming and often produce off various polluting gases, and conventional precipitation and photodeposition are difficult to synthesize Ni9S8. Hydrothermal synthesis has attracted much attention because of its simple operation, safety, high efficiency, energy saving and environmental friendliness. However, the conventional hydrothermal methods to prepare the composites of g-C3N4 and nickel sulfides generally involve in complicated synthesis steps and produce a mixture of several nickel sulfides (NiS and NixSy).
Therefore, in the present work, we adopt a facile, green, one-step hydrothermal route, which is based on the principle of electroless nickel plating, to grow Ni9S8 nanostructures onto the pre-prepared g-C3N4 nanosheet, obtaining the g-C3N4/Ni9S8 composite [22, 33]. In the synthesis of g-C3N4/Ni9S8 composite, in order to form Ni9S8, NaH2PO2 was used, which is commonly used in electroless nickel plating, to reduce Ni2+ to metal Ni, and then to conduct the redox reaction with S0 and S2− to obtain the composites of g-C3N4 with pure Ni9S8. Compared with the traditional hydrothermal methods, the reduction and sulfuration of Ni2+ were concentrated in one-step hydrothermal synthesis, much reducing the processes. Moreover, the as-acquired g-C3N4/Ni9S8 composite has a uniform dispersion of Ni9S8 nanostructures on the g-C3N4 nanosheet. Because it possesses high-conductivity, low surface work function and high electron mobility, the as-acquired composite photocatalyst has excellent photocatalytic performance for hydrogen evolution, presenting a H2 evolution rate as high as 355.7 mmol g−1 h−1 at 7 °C. The successful preparation of the present composite will also provide a new perspective for developing other high-performance heterostructure photocatalysts for hydrogen evolution.
2 Experimental
2.1 Raw materials
Sodium hypophosphite (NaH2PO2, 99.0%), nickel acetate [Ni(CH3COO)2, 99.5%], melamine (99.5%), thiourea (CH4N2S, 99.5%), ethanol (C2H5OH, 99.9%), triethanolamine (TEOA, 99.5%), H2PtCl6·6H2O (99.5%) and Na2SO4 (99.5%) were bought from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). During the experiments, all the raw materials are used without further treatment.
2.2 Synthesis of g-C3N4 particles
Bulk g-C3N4 samples were first synthesized by directly heating melamine [34]. Typically, in an open box furnace, a half-covered crucible with 5 g of melamine powder is heated from room temperature to 550 °C at a speed of 5 °C min−1, soaking at this temperature for 4 h. Afterwards, the furnace was cooled down to ambient temperature, and then the resultant bulk sample was ground, finally obtaining yellow g-C3N4 powders.
2.3 Synthesis of g-C3N4/Ni9S8 nanocomposites
A facile one-step hydrothermal approach was applied in this work to synthesize the proposed g-C3N4/Ni9S8 nanocomposites. Typically, in a beaker with 50 mL deionized water, 100 mg (1.111 mmol) of the as-obtained g-C3N4 powders was homogeneously dispersed through vigorous stirring for 30 min under ultrasonicating. Then, in order to obtain g-C3N4/Ni9S8 composites with different loading amounts of Ni9S8, a series of designed feed amount of nickel acetate (0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.7 and 0.9 mmol, respectively) were added into the dispersion solution. And thiourea with an amount of three times that of nickel acetate was mixed in the dispersion solution together. After further strong stirring for 30 min, 300 mg sodium hypophosphite was mixed in the solution system. After homogeneous mixing, the solution was transferred into an autoclave (100 mL) and then kept in an oven at 140 °C for 8 h. By simply shutting the electricity of the furnace, it was cooled down to ambient temperature. And then the autoclave was taken out from the oven. The resultant precipitates were collected after centrifuging, washed twice with 30 mL deionized water as well as absolute ethanol, respectively, and dried out at 60 °C for 8 h. Finally, the proposed, dark blue powders could be acquired.
2.4 Synthesis of g-C3N4/Pt nanocomposite
The g-C3N4/Pt nanocomposite was prepared by a modified photodeposition method [35]. Typically, 50 mg g-C3N4 was firstly dispersed into 100 mL of 10 vol.% TEOA aqueous solution. After 30 min of ultrasonic dispersion, the solution was transferred to a photoreaction cell. And then 1 wt% chloroplatinic acid (in aqueous solution) was added, compared with the amount of g-C3N4. After that, the air in the reaction system was pumped off, and then a light irradiation was carried out with a 300 W Xe lamp for 1 h to ensure that Pt could be fully deposited on the surface of g-C3N4. Finally, after pumping out the hydrogen produced during the deposition of Pt, the test on photocatalytic hydrogen evolution over the as-prepared g-C3N4/Pt nanocomposite was directly carried out.
2.5 Materials characterization
In this work, a Zeiss GEMINISEM 500 field emission scanning electron microscope (FE-SEM, Germany) and JEOL JEM-2100F transmission electron microscope (TEM, Japan) were applied to examine the morphology and microstructure of the samples. Then a Thermo ESCALAB MKII X-ray photoelectron spectroscope (XPS, Thermo VG Scientific Ltd., UK) was used to explore their elemental composition and chemical state. With the C1s line (284.8 eV) as reference, the recorded results were corrected. In order to identify their phase compositions, X-ray diffraction (XRD) analysis was executed (GI-XRD, Japan; Cu Kα radiation, λ = 1.5418 Å). For the analysis, the corresponding scanning rate was set as 4° min−1 with 1° of X-ray incidence angle in continuous scanning mode. The nitrogen adsorption–desorption isotherms were recorded on a Quantachrome Autosorb-iQ Adsorption–Desorption Instrument (America). The surface area and corresponding pore size distribution were obtained by the Brunauer–Emmette–Teller (BET) and Barrett–Joyner–Halenda (BJH) method, respectively. In order to judge the charge transfer efficiency, photoluminescence spectra were recorded on a FLS980 fluorometer (PL, Edinburgh Instrument, England) at an excitation wavelength of 320 nm. Finally, with a Varian Cary 5000 UV–Vis spectrometer (Agilent, America), the UV–visible absorption spectrum of the samples was collected.
2.6 Evaluation of photocatalytic hydrogen evolution
To acquire the accurate data of hydrogen evolution, in each test 50 mg catalyst was first diffused into 100 ml TEOA aqueous solution (10 vol.%). Then, the prepared reaction mixture was settled in a Perfectlight LabSolar PhotocatalyticHydrogen Evolution System (Beijing, China). The system possesses a Xe lamp (300 W) with an UV cut-off filter (λ ≥ 420 nm) for light source. Afterwards, the system was sealed. Before photocatalytic reaction, the system was first evacuated to a vacuum of − 0.1 MPa, and throughout the photocatalytic reaction, the cooling system based on circulating water should work continuously to keep the reaction temperature at 7 °C. For the reaction, the applied light source was placed 20 cm far from the reaction vessel, and the area for effective irradiation was measured as 12.57 cm2. During the reaction, the generated gas was characterized by a gas chromatography (GC-7900, Xuansheng Scientific Instrument Co. Ltd, Shanghai, China) on-line with nitrogen as carrier gas. Additionally, each of the cyclic tests for photocatalytic hydrogen evolution was executed for 3 h and the gas products were taken for characterization every half an hour. After one round of tests, the reactor will be evacuated for 30 min, and then the subsequent round of tests is repeated without changing the reaction liquid. Totally 6 rounds of tests were performed to evaluate the cyclic performance.
2.7 Photoelectrochemical characterization
For each photoelectrochemical test, a working electrode was first fabricated, for which 50 μL ink prepared from 5 mg sample, 500 μL absolute ethanol together with 20 μL Nafion solution (5 wt%) was coating onto a FTO glass substrate, finally forming an electrode with a size of 1 cm2. The electrolyte is 0.2 M Na2SO4 aqueous solution.
The measurement on transient photocurrents was carried out by a standard three-electrode system on an electrochemical workstation (CHI 660E, Chenhua Instrument, Shanghai, China). For the measurement, a piece of Pt foil was used as counter electrode, Ag/AgCl (saturated KCl) was used as reference electrode, and the visible light (λ ≥ 420 nm) was provided by a Xe lamp (300 W) using an UV cut-off filter. Prior to the tests, the system should be degassed by high-purity N2 gas for about 30 min.
The electrochemical impedance spectra (EIS) of the samples were recorded with the same parameters as those in measuring transient photocurrents. During the EIS tests, the frequency falls in the range of 0.01–100,000 Hz, and the applied AC amplitude is 5 mV (vs. Ag/AgCl).
3 Results and discussion
3.1 Photocatalytic performance for H2 evolution
To reveal the photocatalytic activity of the proposed g-C3N4/Ni9S8 composite, all the samples were tested for hydrogen evolution under the irradiation of visible light at 7 °C. Firstly, a series of samples prepared with different feed molar ratios of Ni2+/g-C3N4 were examined to determine the optimal sample. Figure 1a compares their hydrogen evolution rates (HERs). As can be seen in this figure, with the increase of feeding amount of nickel source, the HER value of the acquired samples can be improved. When the Ni2+/g-C3N4 feed molar ratio was 0.18, their HER value reached the highest, which was 355.7 μmol g−1 h−1. With more nickel source added, the performance of the obtained photocatalysts declined again, possibly because the excessive addition of nickel source would lead to a so large amount of nickel sulfides coating on g-C3N4 (see Table 1), which would hinder the absorption of light by g-C3N4, thus reducing the hydrogen evolution efficiency of the catalysts. So, the optimal Ni2+/g-C3N4 feed molar ratio for the present composite samples was determined as 0.18. In Fig. 1b, the HER values of the raw g-C3N4, the pure Ni9S8, the raw g-C3N4 which loaded with Pt (1 wt%) and the optimal g-C3N4/Ni9S8 composite are compared. Among them, the sample of g-C3N4 with Pt was directly tested after the deposition of Pt onto g-C3N4 without any purification (see Sect. 2.4). As is seen in the figure, the sample of raw g-C3N4 only has a very small HER and the pure Ni9S8 has no detectable photocatalytic hydrogen evolution ability. However, the HER value of the optimal composite sample can reach 355.7 μmol g−1 h−1. This remarkable value is 21.2 times that of the raw g-C3N4, and only 22.1% less than that of the sample of g-C3N4 loaded with 1 wt% Pt. Therefore, it can be inferred that the compositing between g-C3N4 and Ni9S8 will present a synergistic effect, which can effectively transfer the photogenerated electrons on g-C3N4 to Ni9S8 through the interface charge transfer effect. And then the electrons on Ni9S8 can also combine with H+ to generate hydrogen, which greatly improves the photocatalytic efficiency of g-C3N4. Additionally, in order to determine the stability of the sample, the cycling tests were carried on the optimal g-C3N4/Ni9S8 composite. For the cycling tests, the total time was 18 h, which was performed in six rounds and each round was carried out for 3 h in the light reaction cell. For accurate measurement, at the end of each round of test, the hydrogen in the system was evacuated by pumping off and then a new round of test was executed. It can be seen from Fig. 1c that the HER value of the optimal sample has no obvious change after 18 h of cyclic test. Therefore, it can be concluded that the optimal composite has good cycling stability.

Photocatalytic performance on hydrogen evolution: a of the composites prepared with different feed molar ratios of Ni2+/g-C3N4, b comparison on raw g-C3N4, pure Ni9S8, 1 wt% Pt coated g-C3N4 by light deposition and the optimal g-C3N4/Ni9S8 sample, and c the cycling tests on the optimal g-C3N4/Ni9S8 composite. The photocatalytic reactions were carried out with 20 mg catalyst, 100 mL aqueous solution containing 10 vol.% TEOA and a 300 W Xe lamp with UV cut-off filter (λ ≥ 420 nm)
3.2 Compositional and structural properties
To elucidate the photocatalytic performance of the acquired samples, their composition and structure were investigated. Firstly, SEM imaging was carried out. Figure 2a shows typical SEM micrograph of the optimal g-C3N4/Ni9S8 composite. As is seen, this composite sample has a similar morphology with the raw g-C3N4 sample (see Fig. 2b). Therefore, it can be deduced that the two-dimensional structure of raw g-C3N4 is not destroyed during the processing. In order to further examine the microstructure of the composite, TEM imaging was performed on the optimal sample. From the low-resolution image (Fig. 2c), it can be observed that the optimal sample has a stacked lamellar structure, and some black blocks are located on the lamellar structure, which might be Ni9S8 nanostructures loading on g-C3N4. To further confirm the loading of Ni9S8 on g-C3N4, typical high-resolution image is presented in Fig. 2d. From this picture, it can be seen that the clear lattice fringes (0.28 nm), corresponding to the (311) crystalline plane of Ni9S8, and the amorphous area (g-C3N4) are connected closely with each other, most possibly forming heterostructures between g-C3N4 and Ni9S8. In addition, to explore the distribution of nickel sulfides in the sample, EDS mapping scanning was executed in the same area for the low-resolution TEM image. And the results are displayed in Fig. 2e–h. It is easily seen that C, N, Ni as well as S atoms are distributed over the sampling area. Therefore, it can be also concluded that the Ni9S8 nanostructures in the obtained composite are uniformly grown onto the g-C3N4 surface, rather than a simple mixture of both components, indirectly confirming the heterostructures between g-C3N4 and Ni9S8. The uniform composition and structure are one of the reasons why the proposed g-C3N4/Ni9S8 composite has high photocatalytic activity.

Microstructure and mainly elemental distribution. SEM images of the optimal sample (a) and g-C3N4 (b). TEM images of the optimal sample: c low-resolution, d high-resolution. And TEM-EDS mapping images of e C, f N, g Ni, and h S
To determine the phase composition of the specimens, XRD analysis was carried out. Figure 3 compares the recorded XRD patterns of the optimal composite, the raw g-C3N4 and pure nickel sulfides samples, in which the raw g-C3N4 specimen was acquired by the calcination of melamine, while the nickel sulfides sample was obtained under the similar conditions as done for the optimal composite but without the addition of g-C3N4. As is seen, the main XRD peaks of the pure nickel sulfides sample can be indexed to orthorhombic Ni9S8 phase (JCPDS No. 22-1193), while a small amount of hexagonal NiS (JCPDS No. 12-0041) can be also identified, indicating that it is a mixture of Ni9S8 as the main body with a small amount of hexagonal NiS. The XRD peaks of the raw g-C3N4 sample match perfectly with those of its predecessors, confirming its successful synthesis in this work [13]. And in the optimal composite, only the diffraction peaks of g-C3N4 and Ni9S8 can be identified, which proves that it is a relatively pure composite of g-C3N4 and Ni9S8.

XRD patterns of the optimal g-C3N4/Ni9S8 composite, g-C3N4 and pure nickel sulfides samples
The elemental composition as well as chemical state of the optimal g-C3N4/Ni9S8 composite was further detected by XPS analysis. The obtained results are shown in Fig. 4 and Table 1. XPS survey spectroscopy reveals that the g-C3N4/Ni9S8 samples are composed of C, N, Ni and S, indicating that they are a composite of these elements as expected (see Fig. 4a). As for the peak of O element, it might be caused by the oxidation of g-C3N4 and/or the coupling of hydroxyl groups on the samples during hydrothermal synthesis [24]. Moreover, with increasing feed molar ratio of Ni2+/g-C3N4, the loading amount of Ni9S8 on g-C3N4 increased (Table 1). The high-resolution C 1 s spectrum as exhibited in Fig. 4b presents two distinct peaks at 284.75 and 287.91 eV. Literature survey reveals that the peak at 284.75 eV could be assigned to free carbon, while the one at 287.91 eV should be indexed to the binding energy of C atoms in g-C3N4 [36]. The N 1 s spectrum (see Fig. 4c) can be fitted into four peaks at 398.36, 399.03, 400.69 and 404.25 eV, respectively. Among them, the three peaks at 398.36, 399.03 and 400.69 eV could be ascribed to the sp2 hybrid nitrogen in C=N–C group, sp3 hybrid nitrogen in N–(C)3 structure and nitrogen in amino moiety in g-C3N4, respectively, while the one at 404.25 eV is owing to the π-excitation of N in the g-C3N4 structures [37]. And the binding energy of C 1s and N 1s in the g-C3N4/Ni9S8 composite has no obvious change compared with that of the pure g-C3N4. So it can be deduced that the valence state of g-C3N4 is not changed after their combination. The Ni 2p3/2 spectrum (Fig. 4d) exhibits three main peaks at 853.15, 855.83 and 861.29 eV, in which the peaks at 853.15 and 855.83 eV can be attributed to Ni2+ in Ni9S8, and that at 861.29 eV is assigned to Ni2+ in the hydroxyl compound of nickel [27]. However, as is mentioned above, the phase of hydroxyl compound of nickel could not be identified from the XRD patterns, possibly because its content in the samples was too small. In a word, the main reason for the formation of this binding energy is the hydroxyl hanging bond on the surface of Ni9S8 during hydrothermal reaction [10]. Correspondingly, the S 2p spectrum presented in Fig. 4e displays three main peaks at 161.92, 163.08 and 168.63 eV. The peaks at 161.92 and 163.08 eV can be attributed to the S2− and S22− in Ni9S8 lattice, while that at 168.63 eV may be due to the attachment of some sulphate produced by thiourea hydrolysis on the surface of the sample [27]. At the same time, compared with pure Ni9S8, the binding energies of Ni 2p and S 2p in the g-C3N4/Ni9S8 composite are slightly shifted to high energy, indicating that the electrons in g-C3N4 are transferred to Ni9S8, which also confirms the combination between g-C3N4 and Ni9S8.

XPS results of the optimal g-C3N4/Ni9S8 composite. a Survey spectrum. High resolution spectra of b C 1s, c N 1s, d Ni 2p and e S 2p. For comparison, the corresponding data of pure g-C3N4 or Ni9S8 samples are also displayed in (b–e)
To obtain the specific surface area (SBET), pore size distribution and pore volume of the optimal g-C3N4/Ni9S8 composite, low-temperature nitrogen adsorption tests were carried out. Figure 5a presents the nitrogen adsorption–desorption isotherms in comparison with that of pure g-C3N4. Their corresponding pore size distribution curves were obtained by the multipoint BET and BJH methods, and the results are shown in Fig. 5b. The calculated SBET, pore volume and pore size of the raw g-C3N4 and g-C3N4/Ni9S8 composite are listed in Table 2. From Table 2, it can be seen that the specific surface area of the optimal g-C3N4/Ni9S8 composite became smaller after the compositing of g-C3N4 with Ni9S8. And from Fig. 5b, it can be observed that after compositing, the pore volume of the optimal g-C3N4/Ni9S8 composite at the pore size of about 40 nm significantly decreased, but that at the pore size of 4 nm changed very little. These results indicate that, the growth of Ni9S8 happens in the large pores of g-C3N4, and the deposition of Ni9S8 will have a certain blockage effect on the pores of g-C3N4. Therefore, it can be concluded that the improved catalytic performance of the optimal g-C3N4/Ni9S8 composite is not attributed to the increased number of active sites originating from the enhancement in specific surface area of the composite, but to the prompt transfer of electrons instead caused by the heterostructured combination between g-C3N4 and Ni9S8.

BET results: nitrogen adsorption–desorption isotherms (a) and the corresponding pore size distribution curves (b) of the optimal g-C3N4/Ni9S8 composite and the raw g-C3N4
Figure 6a compares the UV-visible absorption spectrum of the optimal g-C3N4/Ni9S8 composite with those of the raw g-C3N4 and Ni9S8 nanostructure. It can be seen that the starting point of the absorption edge for the raw g-C3N4 is about 440 nm, which is well corresponding to the value reported in literature [38]. However, after g-C3N4 was combined with Ni9S8, the absorption ability for visible light of the obtained composite was greatly enhanced, while the absorption capacity for ultraviolet and near ultraviolet spectrum below 450 nm was also significantly increased. This result reveals that after the Ni9S8 co-catalyst was loaded onto the g-C3N4 nanosheets, the optical absorption region of the composite sample could be effectively broadened. The enhanced ability in optical absorption could be owing to the existence of low bandgap black Ni9S8 in the g-C3N4/Ni9S8 composite. Such Ni9S8 nanostructures have strong optical absorption to the light with a wavelength from 300 to 800 nm. Furthermore, the bandgaps of the optimal g-C3N4/Ni9S8 composite and the raw g-C3N4 sample were estimated from their corresponding plots on (αhν)2 versus Eg (see Fig. 6b). As is seen, the Eg values of the optimal g-C3N4/Ni9S8 composite and the raw g-C3N4 are 2.85 and 2.88 eV, respectively. In order to further explore the band gap structure of the present photocatalyst materials, the density of states (DOS) of the valence band of the optimal g-C3N4/Ni9S8 composite and raw g-C3N4 were measured by valence band XPS (Fig. 6c). Both of them displayed typical valence band DOS characteristics of g-C3N4 with the edge of the maximum energy at about 0.07 and 0.34 eV, respectively. According to the band gap obtained from the UV spectrum, it can be calculated that the conduction band of the optimal g-C3N4/Ni9S8 composite and the raw g-C3N4 would occur at about − 2.87 and − 2.51 eV, respectively. Therefore, it can be inferred that thermodynamically, the g-C3N4/Ni9S8 composite has a better hydrogen evolution ability due to its more negative conduction band. The interfacial charge transfer efficiency of the optimal g-C3N4/Ni9S8 and raw g-C3N4 can be evaluated from the stable photoluminescence (see Fig. 6d). The raw g-C3N4 has a strong photoluminescence peak at 470 nm. However, compared with the raw g-C3N4, the photoluminescence peak intensity of the optimal g-C3N4/Ni9S8 composite becomes significantly weaker. These results indicate that the present g-C3N4/Ni9S8 has a higher charge transfer efficiency, which can be ascribed to the fast transport of photoelectrons from g-C3N4 to Ni9S8, thus finally improving the separation efficiency of photogenerated carriers. In addition, the photoluminescence peak of g-C3N4/Ni9S8 shifts from 470 to 462 nm in comparison with that of the raw g-C3N4, which also confirms the loading of Ni9S8 on g-C3N4 in the composite samples.

Optical properties. a UV-Vis absorption spectra of the optimal g-C3N4/Ni9S8 composite, raw g-C3N4 and Ni9S8, and b the corresponding (αhν)2 versus Eg plots. c Valence-band XPS spectra and d stable photoluminescence spectra of the optimal g-C3N4/Ni9S8 composite and raw g-C3N4
In summary, the above-mentioned characterizations reveal that a g-C3N4/Ni9S8 composite with uniform composition and structure has been successfully synthesized in this work.
Finally, the formation mechanism of the present g-C3N4/Ni9S8 composite as follows was proposed based on the above experimental results (see Fig. 7). In the designed first step, the raw g-C3N4 powder is dispersed in deionized water by ultrasonic stirring to form uniformly dispersed g-C3N4 nanosheets. After nickel acetate and thiourea joined into the reaction system, because of the negative charges on the g-C3N4 nanosheets, Ni2+ could be adsorption on the surface of g-C3N4, while thiourea could form a complex with Ni2+ to slow down the release of Ni2+. In the second step, during the hydrothermal reaction, sodium hypophosphite was first decomposed into PH3, which could be ionized into H− with strong reducibility. Then the H− can reduce Ni2+ attached to g-C3N4 surface into metal Ni. In the third step, the stable thiourea was thermally decomposed to produce a large amount of S2−, which would react with the metal Ni on the g-C3N4 surface to obtain the proposed composite in the state full of unsaturated sulfur. Finally, the g-C3N4/Ni9S8 composite in the state of unsaturated sulfur was obtained.

Synthesis mechanism of the present g-C3N4/Ni9S8 composite
3.3 Photocatalytic mechanism
To clarify the photocatalytic mechanism for H2 evolution of the as-acquired g-C3N4/Ni9S8 composite, its photoelectrochemical features were further explored. For comparison, the Ni9S8 nanostructure and raw g-C3N4 were also investigated. Firstly, the transient photocurrents of the raw g-C3N4, pure Ni9S8 and the optimal g-C3N4/Ni9S8 composite are compared in Fig. 8a. It can be seen from this figure that the raw g-C3N4 sample has only 0.3 μA cm−2 of transient photocurrent, which is far less than that of the optimal composite. This is because the photogenerated electrons and holes are easily recombined with each other in the raw g-C3N4 sample due to its low-conductivity. In addition, on the pure Ni9S8 nanostructure the detected photocurrent is not distinct, indicating that although it has strong absorption to the light at the wavelength of 300–800 nm, it cannot contribute to the transient photocurrent of the present composite independently. These facts prove that in the present g-C3N4/Ni9S8 composite, the Ni9S8 nanostructure is not a photocatalyst but a co-catalyst for g-C3N4. The Ni9S8 co-catalyst can boost the segregation of photogenerated electrons and holes, thus increasing the transfer efficiency of photogenerated carriers.

Photoelectrochemical data of the optimal g-C3N4/Ni9S8 composite, raw g-C3N4 and pure Ni9S8 nanostructure: a transient photocurrent responses, and b EIS Nyquist plots
Furthermore, the recorded EIS Nyquist curves (see Fig. 8b) reveal that among the three investigated samples, the raw g-C3N4 sample possesses the highest intrinsic impedance, indicating that there is a higher charge transfer rate in the composite g-C3N4/Ni9S8 samples after the loading of Ni9S8 onto g-C3N4. These phenomena may be due to the homogeneous combination of the relatively low-conductivity g-C3N4 nanosheets with the high-conductivity Ni9S8 nanostructures.
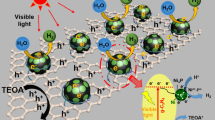
According to the above-mentioned experimental results, the possible photocatalytic mechanism over the present g-C3N4/Ni9S8 composite on hydrogen evolution was proposed (see Fig. 9). Based on the calculated positions of conduction band and valence band of g-C3N4/Ni9S8 composite, under the irradiation of visible light, the photogenerated electron (e−) will leap into the conduction band of g-C3N4, which will leave hole (H+) in its valence band. A part of photogenerated electrons could move to the g-C3N4 surface and directly reduce H+ ions in solution to produce H2. More importantly, due to the close contact in the heterostructure between the main body g-C3N4 and the high-conductivity Ni9S8 nanostructures in the present g-C3N4/Ni9S8 composite, a built-in electric field is formed after the combination between them. As a result, the photogenerated electrons can easily emigrate to the Ni9S8 nanostructures, thus effectively suppressing the recombination between the photogenerated electrons and holes. Because of the low impedance and high electrocatalytic activity of Ni9S8, the photogenerated electrons transferred to Ni9S8 can rapidly move to the surface of Ni9S8, which can also serve as the active site for electron reduction reaction, effectively reducing H+ to H2 in the solution. In addition, triethanolamine can absorb the holes transferred from the valence band to the surface of g-C3N4, thus completing a whole set of redox reactions.

Photocatalytic mechanism for hydrogen evolution over the optimal g-C3N4/Ni9S8 composite
4 Conclusions
An interesting g-C3N4/Ni9S8 composite was prepared using a novel hydrothermal method, in which nickel acetate and thiourea were used as the Ni and S sources, respectively, while NaH2PO2 was used as the reducing agent, finally synthesizing the g-C3N4/Ni9S8 composite through a simple one-step process. This method has the potential for large-scale production of g-C3N4/Ni9S8 composite with the advantages of simple, safety, environmental friendliness and easy controllability. The optimal g-C3N4/Ni9S8 nanocomposite has high photocatalytic activity under visible light irradiation. The hydrogen evolution rate of the optimal g-C3N4/Ni9S8 composite prepared with the Ni2+/g-C3N4 feed molar ratio of 0.18 could reach 355.7 μmol g−1 h−1 at 7 °C, which is 21.2 times higher than that of the pure g-C3N4 sample, and only 22.1% less than that of the g-C3N4 with Pt (1 wt%). The greatly enhanced performance on photocatalytic hydrogen evolution over the as-acquired g-C3N4/Ni9S8 composite could be attributed to the prompt transfer of photogenerated electrons from the low-conductivity g-C3N4 surface to the high-conductivity Ni9S8 cocatalyst, which effectively promotes the segregation of photogenerated electrons and holes, and sponsors the electrocatalytic activity of Ni9S8 via effectively reducing H+–H2 as well. The present study will provide a new approach for the construction of photocatalysts by co-catalysis for photocatalytic hydrogen evolution.
References
V. Ramanathan, M.V. Ramana, G. Roberts, D. Kim, C. Corrigan, C. Chung, D. Winker, Warming trends in Asia amplified by brown cloud solar absorption. Nature 448, 575-U5 (2007)
G. Glenk, S. Reichelstein, Economics of converting renewable power to hydrogen. Nat. Energy 4, 212–216 (2019)
A. Fujishima, K. Honda, Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37–38 (1972)
L. Buzzetti, G.E.M. Crisenza, P. Melchiorre, Mechanistic studies in photocatalysis. Angew. Chem. Int. Ed. 58, 3730–3747 (2019)
Y.R. Li, J. Xu, Z.Y. Liu, H. Yu, Performance of amorphous CoSx/oxygen vacancies ZnO heterojunction photocatalytic hydrogen evolution. J. Mater. Sci. Mater. Electron. 30, 246–258 (2020)
X. Wang, Q. Xu, M.R. Li, S. Shen, X.L. Wang, Y.C. Wang, Z.C. Feng, J.Y. Shi, H.X. Han, C.P. Li, Photocatalytic overall water splitting promoted by an alpha-beta phase junction on Ga2O3. Angew. Chem. Int. Ed. 51, 13089–13092 (2012)
S.F. Wang, H.J. Gao, X.L. Yu, S.N. Tang, Y. Wang, L.M. Fang, X.X. Zhao, J.Y. Li, L. Yang, W.Q. Dang, Nanostructured SrTiO3 with different morphologies achieved by mineral acid-assisted hydrothermal method with enhanced optical, electrochemical, and photocatalytic performances. J. Mater. Sci. Mater. Electron. 31, 17736–17754 (2020)
P. Zhang, B.Y. Guan, L. Yu, X.W. Lou, Facile synthesis of multi-shelled ZnS-CdS cages with enhanced photoelectrochemical performance for solar energy conversion. Chem 4, 162–173 (2018)
S.V.P. Vattikuti, P.A.K. Reddy, J. Shim, C. Byon, Visible-light-driven photocatalytic activity of SnO2-ZnO quantum dots anchored on g-C3N4 nanosheets for photocatalytic pollutant degradation and H2 production. ACS Omega 3, 7587–7602 (2018)
S.D. Guan, X.L. Fu, Y. Zhang, Z.J. Peng, β-NiS modified CdS nanowires for photocatalytic H2 evolution with exceptionally high efficiency. Chem. Sci. 6, 1574–1585 (2018)
G. Mahalakshmi, M. Rajeswari, P. Ponnarasi, Fabrication of dandelion clock-inspired preparation of core-shell TiO2@MoS2 composites for unprecedented high visible light-driven photocatalytic performance. J. Mater. Sci. Mater. Electron. 31, 22252–22264 (2020)
D. Chen, Z.F. Liu, Z.G. Guo, W.G. Yan, M.N. Ruan, Decorating Cu2O photocathode with noble-metal-free Al and NiS cocatalysts for efficient photoelectrochemical water splitting by light harvesting management and charge separation design. Chem. Eng. J. 381, 122655 (2020)
W.J. Ong, L.L. Tan, Y.H. Ng, S.T. Yong, S.P. Chai, Graphitic carbon nitride (g-C3N4) based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016)
X.C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J.M. Carlsson, K. Domen, M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80 (2009)
D. Masih, Y.Y. Ma, S.G. Rohani, Graphitic C3N4 based noble-metal-free photocatalyst systems: a review. Appl. Catal. B 206, 556–588 (2017)
C. Prasad, H. Tang, Q.Q. Liu, I. Bahadur, S. Karlapudi, Y.J. Jiang, A latest overview on photocatalytic application of g-C3N4 based nanostructured materials for hydrogen production. Int. J. Hydrog. Energy 45, 337–379 (2020)
K.C. Devarayapalli, K. Lee, H.-B. Do, N.N. Dang, K. Yoo, J. Shim, S.V.P. Vattikuti, Mesostructured g-C3N4 nanosheets interconnected with V2O5 nanobelts as electrode for coin-cell-type-asymmetric supercapacitor device. Mater. Today Energy 21, 100699 (2021)
S.V.P. Vattikutia, B.P. Reddy, C. Byon, J. Shim, Carbon/CuO nanosphere-anchored g-C3N4 nanosheets as ternary electrode material for supercapacitor. J. Solid State Chem. 262, 106–111 (2018)
L.J. Zhang, X.Q. Hao, Y.B. Li, Z.L. Jin, Performance of WO3/g-C3N4 heterojunction composite boosting with NiS for photocatalytic hydrogen evolution. Appl. Surf. Sci. 499, 143862 (2020)
Z.W. Zhao, Y.J. Sun, F. Dong, Graphitic carbon nitride based nanocomposites: a review. Nanoscale 7, 15–37 (2015)
X. Li, J.G. Yu, M. Jaroniec, X.B. Chen, Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019)
Y. Zhang, Z.J. Peng, S.D. Guan, X.L. Fu, Novel beta-NiS film modified CdS nanoflowers heterostructure nanocomposite: extraordinarily highly efficient photocatalysts for hydrogen evolution. Appl. Catal. B 224, 1000–1008 (2018)
J.H. Lee, S.I. Kim, S.M. Park, M. Kang, A p-n heterojunction NiS-sensitized TiO2 photocatalytic system for efficient photoreduction of carbon dioxide to methane. Ceram. Int. 43, 1768–1774 (2017)
K.L. He, J. Xie, M.L. Li, X. Li, In situ one-pot fabrication of g-C3N4 nanosheets/NiS cocatalyst heterojunction with intimate interfaces for efficient visible light photocatalytic H2 generation. Appl. Surf. Sci. 430, 208–217 (2018)
M.A. Ashraf, Y.F. Yang, A. Fakhri, Synthesis of NiS-MoO3 nanocomposites and decorated on graphene oxides for heterogeneous photocatalysis, antibacterial and antioxidant activities. Ceram. Int. 46, 8379–8384 (2020)
S.D. Guan, X.L. Fu, Z.Z. Lao, C.H. Jin, Z.J. Peng, NiS-MoS2 Hetero-nanosheet arrays on carbon cloth for high-performance flexible hybrid energy storage devices. ACS Sustain. Chem. Eng. 7, 11672–11681 (2019)
A. Khalil, Q. Liu, Z. Muhammad, M. Habib, R. Kha, Q. He, Q. Fang, H.T. Masood, Z. Rehman, T. Xiang, C.Q. Wu, L. Song, Synthesis of Ni9S8/MoS2 heterocatalyst for enhanced hydrogen evolution reaction. Langmuir 33, 5148–5153 (2017)
H. Yang, C. Wang, Y. Zhang, Q. Wang, Chemical valence-dependent electrocatalytic activity for oxygen evolution reaction: a case of nickel sulfides hybridized with N and S co-doped carbon nanoparticles. Small 14, 1703273 (2018)
J.E. Samaniego-Benitez, K. Jimenez-Rangel, L. Lartundo-Rojas, A. Garcia-Garcia, A. Mantilla, Enhanced photocatalytic H2 production over g-C3N4/NiS hybrid photocatalyst. Mater. Lett. 290, 129476 (2021)
J.Q. Wen, J. Xie, H.D. Zhang, A.P. Zhang, Y.J. Liu, X.B. Chen, X. Li, Constructing multifunctional metallic Ni interface layers in the g-C3N4 nanosheets/amorphous NiS heterojunctions for efficient photocatalytic H2 generation. ACS Appl. Mater. Interfaces 9, 14031–13042 (2017)
J.Q. Wen, J. Xie, Z.H. Yang, R.C. Shen, H.Y. Li, X.Y. Luo, X.B. Chen, X. Li, Fabricating the robust g-C3N4 nanosheets/carbons/NiS multiple heterojunctions for enhanced photocatalytic H2 generation: an Insight into the tri-functional roles of nanocarbons. ACS Sustain. Chem. Eng. 5, 2224–2236 (2017)
H. Zhao, H.Z. Zhang, G.W. Cui, Y.M. Dong, G.L. Wang, P.P. Jiang, X.M. Wu, N. Zhao, A photochemical synthesis route to typical transition metal sulfides as highly efficient cocatalyst for hydrogen evolution: from the case of NiS/g-C3N4. Appl. Catal. B 225, 284–290 (2018)
J.J. Huang, C.M. Gui, H.D. Ma, P. Li, W.P. Wu, Z.M. Chen, Surface metallization of PET sheet: fabrication of Pd nanoparticle/polymer brush to catalyze electroless nickel plating. Compos. Sci. Technol. 202, 108547 (2021)
A. Li, Z.J. Peng, X.L. Fu, Exfoliated, mesoporous W18O49/g-C3N4 composites for efficient photocatalytic H2 evolution. Solid State Sci. 106, 106298 (2020)
J.J. Ding, X.X. Sun, Q. Wang, D.S. Li, X.Y. Li, X.X. Li, L. Chen, X. Zhang, X.Y. Tian, K. Ostrikov, Plasma synthesis of Pt/g-C3N4 photocatalysts with enhanced photocatalytic hydrogen generation. J. Alloys Compd. 873, 159871 (2021)
L.F. Cui, X. Ding, Y.G. Wang, H.C. Shi, L.H. Huang, Y.H. Zuo, S.F. Kang, Facile preparation of Z-scheme WO3/g-C3N4 composite photocatalyst with enhanced photocatalytic performance under visible light. Appl. Surf. Sci. 391, 202–210 (2017)
P. Wang, N. Lu, Y. Su, N. Liu, H.T. Yu, J. Li, Y. Wu, Fabrication of WO3@g-C3N4 with core@shell nanostructure for enhanced photocatalytic degradation activity under visible light. Appl. Surf. Sci. 423, 197–204 (2017)
H. Jung, T.T. Pham, E.W. Shin, Interactions between ZnO nanoparticles and amorphous g-C3N4 nanosheets in thermal formation of g-C3N4/ZnO composite materials: the annealing temperature effect. Appl. Surf. Sci. 458, 369–381 (2018)
Acknowledgements
The authors appreciate the financial support for this work from the National Natural Science Foundation of China (Grant Nos. 11674035 and 61274015), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There is no conflict of interests regarding the publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, A., Peng, Z. & Fu, X. Simple hydrothermal synthesis of g-C3N4/Ni9S8 composites for efficient photocatalytic H2 evolution. J Mater Sci: Mater Electron 32, 21643–21657 (2021). https://doi.org/10.1007/s10854-021-06678-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06678-9