
Abstract
Surface self-regeneration of CeO2 nanocomposites based on redox-cycling between Ce3+ and Ce4+ has gingered its wide application and relevance. In this study, rerouted facile green synthesis approach was employed to prepare nanocomposites of CeO2 with chitosan (CS) solutions of ascorbic acid (AAC) and citric acid (CAC). The XRD studies show that pure chitosan, CeO2/CS/AAC, and CeO2/CS/CAC were polycrystalline, with calculated crystallite sizes of 42, 28, and 35 nm, respectively. Meanwhile, CeO2/CS/AAC, and CeO2/CS/CAC were triclinic and hexagonal, respectively. Also, HR-TEM shows spherical nanostructures as well as a globular polycrystalline porous morphology revealed by SEM images. Moreover, the optical properties from UV–Vis spectroscopic studies showed peaks at 214, 292, and 351 nm corresponding to bandgap energies of 5.79, 4.21, and 3.53 eV, respectively. The wide energy bandgap obtained in the study is typical for rare-earth elements and the improved electronic properties of the material is promising for application in electronic/semiconducting devices.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Metal oxide nanocomposites are a class of inorganic nanocomposites that have been extensively studied for many years [1]. Due to the high chemical reactivity of metal ions, they can be transformed into various oxides bearing intriguing physical, chemical, and electronic properties as well as crystallographic forms owing to their small sizes and tunable morphologies [2, 3]. However, the incorporation of rare-earth metal oxide nanocomposites into other systems have gained attention due to their semiconducting properties emerging from the unpaired 5d electron transiting between the lanthanide metal ions and their specific application in electronic devices [4, 5]. Cerium oxide (CeO2) is an imperative n-type semiconductor metal oxide material characterized with interesting properties such as biocompatibility, high isoelectric point, wide bandgap, non-toxicity, brilliant electronic conductivity, and high chemical and electrochemical stabilities [6,7,8]. Additionally, the low temperature of processing, tunable functionalities, optical transparency, chemical inertness, thermal stability, and outstanding redox property between Ce4+ and Ce3+ which promotes high oxygen storage capacity, and oxygen vacancy mobility have chiefly engineered the preparation of CeO2-based nanocomposite [9], which is also helpful in retarding the recombination of photogenerated electron–hole pairs [10, 11]. These unique properties have necessitated the fabrication of nanostructured CeO2 for many applications including sensors [12, 13], fuel cell technology [14, 15], supercapacitors [16], photocatalysts [17] and biomedical fields [18], etc.
The synthesis of polymer/nanocomposite materials is very important in advanced material science as the novel materials combine both the unique properties of the metal precursor and the polymeric material [19], i.e., the polymer matrixes can act as stabilizers/capping agents that prevent aggregation, agglomeration and impact uniform size distribution in the preparation of the metal nanocomposites [20]. Chitosan is a natural cationic biopolymer made up of abundant primary amino (NH2) and hydroxyl (OH) groups in its skeletal backbone and is generally soluble in acidic media [21]. Thus, it is non-toxic and hydrophilic with the special ability to interact with metal ions or other materials to form solutions by ionic/electrostatic interaction, hydrogen bonding, or via metal-chelate complexation reactions [22, 23]. Also, the biopolymer can serve as a good host material for semiconductor nanostructured devices due to its biocompatibility and biodegradability properties and have been widely applied for charge trapping layer in photoinduced systems [24]. Generally, the dissolution of chitosan has been achieved with unpleasant smelling, corrosive and toxic organic acids such as 1–2% glacial acetic acid and acetate buffer solutions, and have been used for the preparation of several CeO2 nanocomposites [24, 25]. For example, Alves et al. [26] used a solution casting approach for the preparation of ionic conducting membranes based on chitosan, glycerol, and cerium triflate Ce(CF3SO3)3, and reported the material as a promising candidate for polymer electrolytes in electrochemical devices. Also, Cai et al. [27] developed an electrochemical immunosensor based on nanocomposite-modified glass carbon electrode by utilizing the biospecific surface of CeO2-chitosan for the determination of sulfamethoxazole in food. In addition, Liang and co-workers [28] synthesized new adsorbent based on mixed rare-earth metals (La and Ce) modified with chitosan for the successful chelation and removal of fluoride ions from drinking water. Some other authors have also reported the preparation of metal oxides based on V2O5, ZrO2, FeO, CuO, TiO2 and MnO2 nanocomposites via different synthesis routes and studied their optical and electrochemical properties for application in numerous fields [29,30,31,32,33,34]. Interestingly, more attention is been focused on the use of natural organic acids (e.g., ascorbic and citric acids) with dual functions of dissolving chitosan and acting as crosslinkers for improving the functional properties of chitosan-based materials [35]. Also, as cerium oxide nanocomposites is often combined with other phases, attempts are made at shaping the final material around a specific hierarchy and with a precise pattern to fine-tune its interaction with other components and fully exploit the corresponding properties [36]. Herein, we have rerouted cerium oxide nanoparticulate embedding by employing a facile green syntheses approach to fabricate a glucose reducing nanostructured cerium oxide composites at ambient conditions. The optical bandgaps and morphological properties of CeO2/CS/AAC and CeO2/CS/CAC nanocomposites were further studied and discussed.
2 Experimental details
2.1 Chemicals
All starting materials (Chitosan, CS (161.1 kDa, cerium nitrate hexahydrate, ascorbic acid (CAS: 50-81-7), citric acid (CAS: 77–92-9), Glucose (99%) and absolute ethanol (99.7%)) with analytic purity were purchased from Maya Reagent, Sinopharm Chemical Reagent Co. Ltd. and Shanghai Macklin Biochemical Co. LTD, China and used without further depuration.
2.2 Synthesis procedure
The synthesis procedure was according to the previous report of [37, 38] with few modifications. Typically, 1.0 g of chitosan was dissolved in 5% (w/v) of ascorbic acid and agitated under a magnetic stirring condition at 60 °C for 45 min to form a clear homogeneous solution. Furthermore, 1.0 mM Ce salt was added with continuous stirring for 60 min. The nanocomposites were formed by the careful addition of 10 mL (1 M) glucose solution gently under stirring for another 60 min until the formation of a pale-yellow precipitate which was centrifuged, washed with ethanol and dried under vacuum at 60 °C overnight. A similar approach was repeated with citric acid until the formation of gray precipitates. The prepared nanocomposites were fully characterized and subjected to optical bandgap test.
2.3 Sample characterization
The as-prepared nanocomposite materials were characterized using Fourier transform infrared (FT-IR) instrument recorded with Nicolet 5700 spectrometer. KBr was used for sample preparation, with data scanning from 4000 to 400 cm−1. Also, the crystallinity of the composites was determined using a Rigaku D/Max-A (Ultima IV) X-ray diffraction (XRD) instrument equipped with a Cu Kα irradiation X-ray source (l = 0.154 nm). The XRD patterns were recorded from 10 to 70° at a scan speed of 20°/min. The thermogravimetric analysis (TGA) profiles of the samples were determined on a NETZSCH TG 209F3 instrument under a nitrogen atmosphere from 30 to 800 °C at a heating rate of 10°/min. Furthermore, the internal morphologies of the nanocomposite were observed using high-resolution transition electron microscopy, HR-TEM (Tecnai G2 T20) at 200 kV voltage, and resolution power of 0.24 nm. The optical absorption properties and electronic transition of the materials were studied using UV-2600 (Shimadzu Corporation, Japan) spectrophotometer with a wavenumber scan from 200 to 800 nm at room temperature. Additionally, the surface morphology of the as-prepared nanocomposites was studied using FEI-110730002486 scanning electron microscope (SEM) coupled with energy dispersive X-ray (EDX) with a magnification of × 20 to 30,000 (Resolution-HV 3.0 nm), while elemental analysis was carried out with an AXIS HIS 165 Spectrometer (Kratos Analytical, Manchester, UK) with a monochromatic Al Kα X-ray source (1486.71 eV photons).
3 Results and discussion
3.1 FT-IR studies
The FT-IR spectra of pure chitosan show broad and weak absorption bands at 3454 cm−1 which is due to the -OH and -NH overlapping stretching vibrations. Also, the band at 1631 cm−1 is assigned to C=O stretching mode in amide I according to previous report [23]. The bands at 1389 cm−1 and 1093 cm−1 are attributed to -NH deformation mode in amide 1 group and C–O–C stretching vibrational mode in glucose circle, respectively (Fig. 1a) [27]. However, the interaction due to the addition of Ce salt was accompanied by hyperchromic shifts (Fig. 1b and c). More so, the shapes of the absorption peaks become intense, broader, and red-shifted due to the involvement of the –OH/–NH overlapping groups in the assembly of CS with CeO2 nanocomposites and subsequent immobilization of the absorbing molecules onto the nanocomposite matrix [39]. Evidently, the marked difference between the peaks due to ascorbic acid and citric acid could be attributed to the different electronic and structural environments of the atoms that make up the compounds, i.e., the conjugated 5-membered ring system in AAC stabilizes and increases the hydrogen bond interaction with CS compared to the competing hydrogen bonding and ionic interaction between the acyclic citrate anions and protonated cationic CS solution (Fig. 2). Thus, the –OH/–NH overlapping band in CS was red-shifted to 3431 cm−1 and 3441 cm−1 due to CS/AAC and CS/CAC, respectively. Also, prominent red shifts were observed due to C=O, C–O–O and -NH deformation bands at 1626 cm−1, 1072 cm−1, and 1382 cm−1 due to CS/AAC and 1590 cm−1, 1086 cm−1 and 1384 cm−1 due to CS/CAC, respectively. It is noteworthy that the stretching band of CeO2 nanoparticles is expected to appear around ~ 465 cm−1 due to Ce–O. In this study, the spectra reveal twin peaks appearing at 617 and 559 cm−1 due to the strong interaction between CS and CeO2 to form the nanocomposite [11].

FT-IR spectra of (a) Pure chitosan (CS) (b) CeO2/CS/AAC nanocomposites (c) CeO2/CS/CAC nanocomposites

Possible (a) Hydrogen bond interaction between CS and conjugated 5-membered ring in AC (b) Ionic interaction between the acyclic citrate anions and protonated cationic CS solution
3.2 X-ray diffraction (XRD) studies
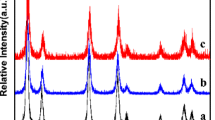
The XRD patterns of chitosan and CS-CeO2 nanocomposites are shown in Fig. 3. According to Fig. 3a, pure chitosan powder shows only one broad peak at 2θ = 19.8o depicting a polycrystalline nature [26]. However, the XRD pattern of the nanocomposites shows reduced peak intensity and reflection planes higher than that of bulk chitosan powder. Bulk or commercially available pure CeO2 crystals are characterized as cubic fluorite crystal facet [40]. Moreover, the XRD pattern of CeO2/CS/AAC nanocomposites (Fig. 3b) shows one very broad diffraction peak at 2θ = 23.6° corresponding to (1 1 1) reflection plane according to JCPDS PDF No. 45–0589, thus revealing a triclinic crystal facet due to cerium nitrate trihydrate (Ce2O(NO3)6.3H2O). The formation of pure polycrystalline CeO2 nanocomposites with hexagonal crystal facets was revealed in the XRD pattern of CeO2/CS/CAC (Fig. 3c) with a single diffraction peak at 2θ = 21.6° corresponding to (1 1 0) reflection plane attributed to JCPDS PDF No. 44-1001, which is in agreement with previous reports of Solanki et al. [41] for chitosan-based CeO2 nanocomposite. Also, the crystallite grain sizes of chitosan and the as-prepared nanocomposite materials were calculated using Debye–Scherrer formula (Eq. 1) [42].

XRD pattern of (a) Pure chitosan powder (b) CeO2/CS/AAC nanocomposites (c) CeO2/CS/CAC nanocomposites
where D is the crystallite size (nm), k is the Debye–Scherrer constant (0.9), λ is the wavelength of the X-ray source (0.15406 nm), β is full width at half maximum, FWHM (radians) and θ is the peak position (radians). The calculated crystallite sizes of the amorphous chitosan powder were 42 nm, while the polycrystalline materials were 28 nm and 35 nm for CeO2/CS/AAC and CeO2/CS/CAC nanocomposites, respectively.
3.3 TGA studies
The TGA studies which reveal the thermal stability of the as-prepared nanocomposite at elevated temperature are presented in Fig. 4. The thermogram of the materials shows three prominent stages of weight loss up to 800 °C. For CeO2/CS/CAC, the first weight loss (6.5%) was observed at 184 °C, while CeO2/CS/AAC show 8.1% weight loss at 95 °C. These weight losses could be attributed to the loss of solvated water molecules in the composite matrix [43]. However, the second stage of total weight losses (44.1% and 56.7%) was observed up to 520 °C and 486 °C for CeO2/CS/CAC and CeO2/CS/AAC, respectively. These losses could be due to the gradual degradation of polymeric chitosan [44]. The third stage of weight loss at higher temperature > 520 °C could be due to the breaking of the intermolecular hydrogen bonds and denaturation of the material [45]. The results show that CeO2/CS/CAC is more thermally stable and could withstand high-temperature conditions with minimal loss in the mass of the material.

Thermogram for TGA of (a) CeO2/CS/AAC (b) CeO2/CS/CAC nanocomposites
3.4 HR-TEM studies
The morphology of the CeO2 nanocomposites investigated by HR-TEM (Fig. 5a–d) shows a spherical-like nanostructure [46]. According to Fig. 5a, the spherical nanostructured particles which are monodispersed could be as a result of the acidity and pH of citric acid solution (3.2) which instigates the easy dispersion of the cerium oxide particles. However, in Fig. 5c, a clustered spherical nanostructured particle was observed. This could be due to the high acidity and lower pH (2.5) of ascorbic acid in solution forcing more dispersion of the cerium oxide particles [47]. Furthermore, the single area electron diffraction (SAED) pattern (Fig. 5b and d) exhibited weak reflections due to the polycrystalline nature of the CeO2 nanocomposites.

HR-TEM micrograph and SAED pattern of (a, b) CeO2/CS/CAC and (c, d) CeO2/CS/AAC nanocomposites, respectively
3.5 SEM–EDX studies
The SEM micrographs of pure CS and the corresponding CeO2/CS/CAC and CeO2/CS/AAC nanocomposites were obtained to show the surface morphologies and interaction between CS and the metal oxide. The SEM micrograph of the pure chitosan powder reveals a smooth surface with some straps which could be due to a high degree of chitin deacetylation (Fig. 6a) [48]. However, the micrographs obtained for the CeO2/CS/CAC and CeO2/CS/AAC nanocomposites displayed a globular polycrystalline porous morphology which implies a strong interaction due to the alteration of the original smooth morphology of pure CS (Fig. 6b, c). This may be attributed to ionic interactions between cationic CS and surface charged CeO2 nanoparticles as observed by Malhotra and Kaushik [11]. Furthermore, elemental mapping of the nanocomposite materials shows an even distribution of C, O and Ce in proportionate amounts (Fig. 7a, b). More so, the EDX spectra of the prepared nanocomposite materials (Fig. 7c, d) show a significant percentage of the elemental composition due to C, O, and Ce with slight variation (Fig. 7c, d inset). However, a remarkably high composition of N was observed in CeO2/CS/CAC which projects the possibility of an ionic interaction in the nanocomposite material. Also, the high percentage composition of oxygen in the materials could be an indication of the redox property between Ce4+ and Ce3+ which can be beneficial in high oxygen storage devices.

SEM micrograph of (a) Pure chitosan powder (b) CeO2/CS/CAC, (c) CeO2/CS/AAC nanocomposites

Elemental mapping of (a) CeO2/CS/CAC and (b) CeO2/CS/AAC nanocomposites. EDX Spectra and percentage elemental composition (Inset) of (c) CeO2/CS/CAC and (d) CeO2/CS/AAC nanocomposites
3.6 XPS studies
The elemental composition and chemical states of the as-prepared cerium oxide nanocomposite were confirmed using XPS with intensity counts from 200 to 1000 eV. The XPS survey scan spectrum shows the major absorption peaks due to C, N, O, and Ce (Fig. 8). The obtained elemental composition and distribution was in agreement with the results obtained from EDX. The XPS spectra reveal an intense peak at 284 eV which corresponds well with the abundance of C 1 s. However, the distribution of N 1 s was revealed by a single peak at 396 eV for CeO2/CS/AAC nanocomposites, while CeO2/CS/CAC nanocomposites displayed two signals at 398 and 496 eV which could be due to N–H and N-metal bonds, respectively. More so, the abundance of O 1 s was revealed in the signal at 533 eV. The XPS spectrum cerium consist of two distinct small signals of Ce 3d5/2 and 3d3/2, corresponding to different ionic states of Ce4+ and Ce3+. These signals appeared at 781 and 975 eV, respectively, representing the relative amount of the cerium’s electronic state concentrations present in the prepared nanocomposites and the spectral data are in tandem with what have been reported in literature for cerium nanocomposites [49, 50].

XPS scan spectrum of (a) CeO2/CS/AAC and (b) CeO2/CS/CAC nanocomposites
3.7 Optical absorption properties
The optical properties of a material can be derived from the energy bandgap which is the energy difference between the valence band (VB) and the conduction band (CB) (measured in electron volts, eV) in insulators and semiconducting devices. Ordinarily, CS is not expected to give rise to UV–Vis absorption because of the absence of chromophoric species in its backbone. Thus, extinction coefficients of CS for wavelengths shorter than approximately 225 nm is non-zero [51]. However, the UV–Vis absorption spectra of the CS/AAC reveal two sharp and intense peaks at 232 nm and 287 nm which could be due to the conjugation in ascorbic acid (Fig. 9a), while that of CS/CAC reveal two peaks: one sharp and intense peak at 230 nm and a weak peak 292 nm (Fig. 9b). Furthermore, the UV–Vis spectra of the as-synthesized CeO2 nanocomposite shown in Fig. 9c and d reveal three prominent hypochromically shifted peaks appearing at around 214 nm, 292 nm, and 351 nm which may be due to interaction between the CS solutions and Ce salt. These electronic absorption could be attributed to n-π* and π-π*electronic transition from O2− 2p to Ce4+ 4f orbital (Eqs. 2 and 3) [43, 52, 53].

UV–Vis spectra of the as-prepared (a) Chitosan ascorbic acid solution (CS/AAC) and (b) Chitosan citric acid solution (CS/CAC). Nanocomposites of (c) CeO2/CS/AAC and (d) CeO2/CS/CAC nanocomposites. Tauc’s plots of (e) CeO2/CS/AAC and (F) CeO2/CS/CAC. g Bandgap energy showing the conduction band and valence band
These absorption peaks correspond to a bandgap energy of 5.79 eV, 4.21 eV and 3.53 eV (typical for CeO2), respectively, according to the relationship (Eq. 4);
where E is the bandgap energy, h is Planck’s constant, c is the speed of light and λ is the wavelength of absorption. However, the experimental photon energies obtained from the Tauc’s plot were 2.09 and 3.11 eV for CeO2/CS/AAC and CeO2/CS/AAC nanocomposites, respectively (Fig. 9e, f). The result shows that CeO2/CS/CAC possess a photon energy equivalent to the bandgap energy for CeO2 materials, which could enable photons to be absorbed by the material and trigger the excitation of an electron into the conduction band. Thus, both minority and majority photon carrier can be generated after absorption [54].
Also, there was a slight increase in the absorbance of CeO2/CS/AAC nanocomposites at 215, 307, 350–376 nm and a significant reduction toward the visible region of the spectra, this could imply that the material will be better suited for visible-light absorption [4]. The is because the corresponding decrease in the bandgap energy from the UV to visible region of the spectra is an implication of the lowering of the energy required for the generation of conduction band electrons (e−) and valence band holes (h+) (Fig. 9g) [55]. This property as well as the wide energy bandgap is typical for rare-earth elements, which is an indication of improved electronic properties suitable for semiconducting devices and supercapacitors [56].
4 Conclusion
CeO2 nanocomposites were successfully prepared via a facile green synthesis route using chitosan solutions of weak organic acids (AAC and CAC) at low temperatures. The as-synthesized nanocomposites were characterized by various techniques to obtain information on its morphology and optical properties. The crystalline phases of the CeO2 were identified by XRD and the calculated crystallite sizes according to Debye–Scherrer equation are given as 42 nm, 28 nm, and 35 nm for pure CS powder, CeO2/CS/AAC, and CeO2/CS/CAC nanocomposites, respectively. However, the formation of CeO2 nanocomposite was confirmed with CeO2/CS/CAC possessing a hexagonal crystal defect and a spherical morphology of the material was revealed by HR-TEM. Additionally, the material shows a wide energy bandgap and improved optical properties, while the results from TGA show that CeO2/CS/CAC could withstand high-temperature conditions with minimal loss in mass of the material. These results obtained from the study are helpful in expanding the syntheses route of CeO2 nanocomposites for application in electronic wares, semiconducting devices, and supercapacitors.
Abbreviations
- CeO2 :
-
Cerium oxide
- CS:
-
Chitosan
- AAC:
-
Ascorbic acid
- CAC:
-
Citric acid
References
G. Manibalan, G. Murugadoss, R. Thangamuthu, P. Ragupathy, R. Mohan Kumar, R. Jayavel, Appl. Surf. Sci. 456, 104 (2018)
M. Fernández-García, A. Martínez-Arias, J.C. Hanson, J.A. Rodriguez, Chem. Rev. 104, 4063 (2004)
G. Murugadoss, R. Jayavel, M. Rajesh Kumar, Superlattices Microstruct. 76, 349 (2014).
F. Zhou, C. Yan, Q. Sun, S. Komarneni, Microporous Mesoporous Mater. 274, 25 (2019)
A.S. Das, D. Biswas, M. Roy, D. Roy, S. Roy, J. Phys. Chem. Solids 124, 44 (2019)
A.A. Ansari, P.R. Solanki, B.D. Malhotra, Appl. Phys. Lett. 92, 2006 (2008)
N.T. Nguyet, L.T.H. Yen, V.Y. Doan, N.L. Hoang, V. Van Thu, H. Lan, T. Trung, V.H. Pham, P.D. Tam, Mater. Sci. Eng. C 96, 790 (2019)
S. Vigneselvan, V. Manikandan, I. Petrila, A. Vanitha, J. Chandrasekaran, J. Phys. Chem. Solids 136, 109173 (2020)
P.K. Labhane, G.H. Sonawane, Inorg. Chem. Commun. 113, 107809 (2020)
M. Jourshabani, Z. Shariatinia, A. Badiei, J. Colloid Interface Sci. 507, 59 (2017)
B.D. Malhotra, A. Kaushik, Thin Solid Films 518, 614 (2009)
G. Manibalan, G. Murugadoss, R. Thangamuthu, R. Mohan Kumar, R. Jayavel, J. Alloys Compd. 773, 449 (2019)
K.R. Jawaher, R. Indirajith, S. Krishnan, R. Robert, S.K.K. Pasha, K. Deshmukh, S.J. Das, J. Sci. Adv. Mater. Devices 3, 139 (2018)
F.S. Sangsefidi, M. Salavati-Niasari, Int. J. Hydrogen Energy 42, 20071 (2017)
M.I. Asghar, S. Jouttijärvi, R. Jokiranta, A.M. Valtavirta, P.D. Lund, Nano Energy 53, 391 (2018)
S. Arunpandiyan, S. Bharathi, A. Pandikumar, S. Ezhil Arasi, A. Arivarasan, Mater. Sci. Semicond. Process. 106, 104765 (2020)
S. Rahemi Ardekani, A. Sabour Rouh Aghdam, M. Nazari, A. Bayat, E. Saievar-Iranizad, M.N. Liavali, Sol. Energy Mater. Sol. Cells 203, 110195 (2019)
C. Maria Magdalane, K. Kaviyarasu, J. Judith Vijaya, B. Siddhardha, B. Jeyaraj, J. Photochem. Photobiol. B Biol. 173, 23 (2017)
N.M. Ushakov, G.Y. Yurkov, L.V. Gorobinskii, O.V. Popkov, I.D. Kosobudskii, Acta Mater. 56, 2336 (2008)
M. A. Ashraf, W. Peng, Y. Zare, K. Y. Rhee, Nanoscale Res. Lett. 13, (2018).
T. Furuike, D. Komoto, H. Hashimoto, H. Tamura, Int. J. Biol. Macromol. 104, 1620 (2017)
S. Adewuyi, I. Cruz-Maya, O. Ejeromedoghene, V. Guarino, in Sustain. Agric. Rev. 36, ed. by G. Crini and E. Lichtfouse, 36th ed. (Springer Nature, 2019), pp. 315–334.
O. Ejeromedoghene, S. Adewuyi, S.A. Amolegbe, C.A. Akinremi, B.A. Moronkola, T. Salaudeen, Nano-Structures and Nano-Objects (2018).
X. He, J. Gan, A. Fakhri, B.F. Dizaji, M.H. Azarbaijan, M. Hosseini, Int. J. Biol. Macromol. 143, 952 (2020)
L. Zhang, T. Zhu, X. Liu, W. Zhang, P. Alimard, Q. Wang, F.F. Gao, X. Zhang, B. Zhang, S. Li, Z. Hu, F.F. Gao, B.D. Malhotra, A. Kaushik, B. Samai, S.C. Bhattacharya, M. Cai, L. Zhu, Y. Ding, J. Wang, J. Li, X. Du, R. Alves, F. Sentanin, R.C. Sabadini, A. Pawlicka, M.M. Silva, P.R. Solanki, C. Dhand, A. Kaushik, A.A. Ansari, K.N. Sood, B.D. Malhotra, X. He, J. Gan, A. Fakhri, B. Faraji, M. Hasani, M. Hosseini, L. Peng, Z. Yi, W. Dongfeng, X.U. Ying, L.U.O. Lan, Polyhedron 518, 1 (2012)
R. Alves, F. Sentanin, R.C. Sabadini, A. Pawlicka, M.M. Silva, Electrochim. Acta 217, 108 (2016)
M. Cai, L. Zhu, Y. Ding, J. Wang, J. Li, X. Du, Mater. Sci. Eng. C 32, 2623 (2012)
P. Liang, Y. Zhang, D. Wang, Y. Xu, L. Luo, J. Rare Earths 31, 817 (2013)
S. Palsaniya, H.B. Nemade, A.K. Dasmahapatra, Carbon N. Y. 150, 179 (2019)
M. Sharma, Nihal, Mater. Today Proc. (2019).
A. Mazabuel-Collazos, C.D. Gómez, J.E. Rodríguez-Páez, Mater. Chem. Phys. 222, 230 (2019)
T.A. Taha, A.A. Azab, Thermal, Optical, and Dielectric Investigations of PVC/La0.95Bi0.05FeO3 Nanocomposites (Elsevier B.V., 2019).
S. Ali, S. Saleem, M. Salman, M. Khan, Mater. Chem. Phys. 122900 (2020).
K. Mohamed Racik, A. Manikandan, M. Mahendiran, P. Prabakaran, J. Madhavan, M. Victor Antony Raj, Phys. E Low-Dimensional Syst. Nanostructures 119, (2020).
P.H. Chen, Y.H. Hwang, T.Y. Kuo, F.H. Liu, J.Y. Lai, H.J. Hsieh, J. Med. Biol. Eng. 27, 23 (2007)
M. Melchionna, A. Trovarelli, P. Fornasiero, Synthesis and Properties of Cerium Oxide-Based Materials (Elsevier Inc., 2020).
F.S. Sangsefidi, M. Salavati-Niasari, M. Ghasemifard, M. Shabani-Nooshabadi, Int. J. Hydrogen Energy 43, 22955 (2018)
X. Fang, H. Song, J. Photochem. Photobiol. B Biol. 191, 83 (2019)
P. Alimard, Polyhedron 171, 98 (2019)
E. Kumar, P. Selvarajan, D. Muthuraj, Mater. Res. 16, 269 (2013)
P.R. Solanki, C. Dhand, A. Kaushik, A.A. Ansari, K.N. Sood, B.D. Malhotra, Sensors Actuators B Chem. 141, 551 (2009)
V. Uvarov, I. Popov, Mater. Charact. 85, 111 (2013)
B. Samai, S.C. Bhattacharya, Mater. Chem. Phys. 220, 171 (2018)
P.-Z. Hong, S.-D. Li, C.-Y. Ou, C.-P. Li, L. Yang, C.-H. Zhang, J. Appl. Polym. Sci. 105, 547 (2007)
Y. Zhang, B. Liu, L. Wang, Y. Deng, S. Zhou, J. Feng, Polymers (Basel). 11, 1 (2019)
S. Usharani, V. Rajendran, Chin. J. Phys. 55, 2588 (2017)
V. Ratchagar, K. Jagannathan, J. Alloys Compd. 689, 1088 (2016)
S. Adewuyi, I.O. Bisiriyu, C.A. Akinremi, S.A. Amolegbe, J. Inorg. Organomet. Polym. Mater. 27, 114 (2017)
M.E. Khan, M.M. Khan, M.H. Cho, Sci. Rep. 7, 1 (2017)
T. Saravanan, M. Shanmugam, P. Anandan, M. Azhagurajan, K. Pazhanivel, M. Arivanandhan, Y. Hayakawa, R. Jayavel, Dalt. Trans. 44, 9901 (2015)
J. Kumirska, M. Czerwicka, Z. Kaczyński, A. Bychowska, K. Brzozowski, J. Thöming, P. Stepnowski, Mar. Drugs 8, 1567 (2010)
N. Ahmadi, A. Nemati, M. Bagherzadeh, J. Alloys Compd. 742, 986 (2018)
S. Ameen, M. Shaheer Akhtar, H.K. Seo, H.S. Shin, Chem. Eng. J. 247, 193 (2014)
B.T. Yuanwen Jiang, Nat. Rev. Mater. 3, 473 (2018)
A.A. Farghali, M.H. Khedr, S.I. El-Dek, A.E. Megahed, Ultrason. Sonochem. 42, 556 (2018)
J.Y. Tsao, S. Chowdhury, M.A. Hollis, D. Jena, N.M. Johnson, K.A. Jones, R.J. Kaplar, S. Rajan, C.G. Van de Walle, E. Bellotti, C.L. Chua, R. Collazo, M.E. Coltrin, J.A. Cooper, K.R. Evans, S. Graham, T.A. Grotjohn, E.R. Heller, M. Higashiwaki, M.S. Islam, P.W. Juodawlkis, M.A. Khan, A.D. Koehler, J.H. Leach, U.K. Mishra, R.J. Nemanich, R.C.N. Pilawa-Podgurski, J.B. Shealy, Z. Sitar, M.J. Tadjer, A.F. Witulski, M. Wraback, J.A. Simmons, Adv. Electron. Mater. 4, 1 (2018)
Acknowledgements
This work was supported by Hydron Contact Lens Co., Ltd (Jiangsu, China) for the development and industrialization of photochromic contact lenses (No. 8507040230).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors hereby declare no known form of competing for financial or personal interest with regard to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ejeromedoghene, O., Oderinde, O., Ma, X. et al. Reroute green synthesis of hexagonal and triclinic nanostructured cerium oxide: morphology and optical properties. J Mater Sci: Mater Electron 32, 16324–16334 (2021). https://doi.org/10.1007/s10854-021-06183-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06183-z