
Abstract
In this paper, using first-principles calculations based on density function theory, we systematically investigate the electronic structures and properties of the bilayer g-SiC3 systems. Among all three bilayer g-SiC3 systems considered here, the AA stacking (all C/Si atoms in upper SiC3 layer lie above the C/Si atoms in lower SiC3 layer) with an indirect bandgap is most stable. The bandgap calculated by PBE and HSE06 are 1.380 eV and 1.959 eV, respectively. Our results revealed that the bandgap could be modulated effectively by applying in-plane biaxial compressing/stretching or vertical strain along the ‘z’ axis. When the tensile stress reaches 3%, the bilayer AA g-SiC3 system changes from semiconductor to semimetal. Moreover, under vertical stretching of ∆d = 0.4 Å (d = 2.83 Å), the bilayer AA g-SiC3 turns from an indirect bandgap semiconductor to a direct bandgap semiconductor, which is attractive for realizing the nanoscale multi-functional device applications. Our results provide a theoretical understanding for future SiC3-based electronic nanodevices with controlled bandgaps.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Since the synthesis of graphene, it has been widely used due to its special electronic properties and potential application in the fields of optoelectronics [1,2,3,4,5]. The use of two-dimensional (2D) materials could be advantageous for a wide range of applications in nanotechnology technology. With the development of graphene, more and more graphene-like materials have been applied to the electronic and photonic devices [6–9], especially silicene and germanene [10,11,12,13,14]. Experimentally, silicene films have been successfully fabricated on metal and semiconductor substrates [12, 13]. Similar to graphene, silicene also has a high carrier mobility [14] due to the linear dispersion relation around the Fermi level. Compared to graphene, the stable silicene has distinct buckling, which is the result of sp2-sp3 hybridization of Si atoms. Moreover, silicene is more preferable in functional room temperature field effect transistor [15]. Recent studies have shown that hybridization is an effective way for achieving desired electronic and optoelectronic properties. Considering the excellent properties of these 2D materials, the researchers are wondering whether the electronic properties could be improved through atomic substitution which is the most direct way of hybridization.
Several studies [14,15,16] have been performed on the compound based on graphene and silicone (known as siligraphenes). The theoretical study on the phase diagrams of three-dimensional Si/C binary systems performed by Gao et al. showing that in addition to the most stable stoichiometry (Si:C = 1:1), there are two metastable structures SiC3 and Si3C [16]. Moreover, compared to the buckling structure of silicene which has sp3-hybridized Si atoms, the planar configurations of g-SiC3 and g-Si3C preserve the sp2 hybridization of C and Si atoms, which avoids the formation of dangling bonds. This not only improves the chemical stability of siligraphenes but also bring the opportunity to facilitate the exfoliation from substrates. At the same time, Tanveer Hussain et al. [17] have demonstrated that g-SiC3 has important applications in the field of lithium battery anode material and photoelectricity. Moreover, SiC3 has strong adsorption capacity in the visible light region, which makes it have a promising application prospect in the field of nanoelectronics and optoelectronics [18,19,20,21]. Encouragingly, C. Pereyra Huelmo et al. have recently reported a new potential synthesis route for SiC3 Siligraphene [22].
In this article, on the basis of the existing studies, the structure and electronic properties of the possible bilayer SiC3 were further studied, including the optimized geometries, lattice constants, binding energies, relative stabilities, energy band structures, density of states and the charge densities. Our results indicate that, by applying the biaxial and vertical strain the energy band structures of the bilayer SiC3 could be controlled effectively, which could lead to appealing character. Our results also have guiding meaning for the experimental synthesis of future SiC3-based electronic nanodevices with controlled bandgaps.
2 Computational methods
We performed all calculations using projection augmented wave (PAW) [23, 24] and generalized gradient approximation (GGA) with Perdew, Burke, and Ernzerhof (PBE) [25] exchange-related functions, implemented in Vienna's scratch simulation package (VASP) [26]. The effect of van der Waals (vdW) interactions is taken into account using the empirical correction scheme of Grimme (DFT-D2/PBE) [27], which has proved to be effective in describing layered structures. A plane-wave basis set with energy cutoff of 800 eV is used to expand the valence electronic wave functions. The supercells are repeated periodically on the x–y plane, while a vacuum region of 20 Å is applied along the ‘z’ direction to avoid spurious interactions between neighboring slabs. The Brillouin zone is sampled with Monkhorst–Pack k point grid of 9 × 9 × 1. Self-consistent solution of Kohn–Sham equation is carried out with the convergence of 10−5 eV. All the structures are fully relaxed until the force on each atom is less than 0.01 eV/Å.
3 Results and discussion
3.1 Electronic properties of monolayer g-SiC3
Using DFT-based calculations, we evaluated the electronic properties of monolayer g-SiC3, including the electronic structures, lattice constants, deformation charge densities, energy band structures and density of states. The calculated results are summarized in Table 1 and Fig. 1. As shown in Fig. 1a, g-SiC3 belongs to the P6/mmm space group. Compared with Graphene, in one hexagonal carbon ring, there are two C atoms replaced by Si atoms in g-SiC3. As we can see from Table 1, our calculated lattice constants ‘a’ of g-SiC3 is 5.630 Å, which agrees well with previous results (5.630 Å in Ref.20, 5.633 Å in Ref.22). In g-SiC3, the length between two C atoms is 1.438 Å, which is slightly larger than the C–C bond length in graphene (1.420 Å) [28].

(a) Atomic structures, (b) deformation charge densities, (c) energy band structures and (d) the total and partial density of states of monolayer g-SiC3 (TDOS and PDOS). The Fermi energy is set as the reference zero energy
Considering that the intrinsic mechanism of these 2D structures is to study the bonding nature between each atom, which actually controls the dynamical behavior of their growth process. We calculated the differential charge density of planes parallel and perpendicular to the atomic layer (shown in Fig. 1b). The deformation charge density \(\Delta \rho\) is defined as the difference between the total charge density in the monolayer and superposition of independent atomic charge densities placed at the atomic sites of the same monolayer, i.e.,
where \(\rho \left(r\right)\) represents the total charge density of the system and \(\sum_{\mu }{\rho }_{atom}\left(r-{R}_{\mu }\right)\) is the superposition of atomic charge densities. The electron accumulation (\(\Delta \rho >0\)) and electron depletion (\(\Delta \rho <0\)) are depicted by orange solid line and blue dashed line, respectively. The result shows that the C–C bonds are strongly covalent, while the C-Si bonds are primarily covalent mixed with partial ionic. Figure 1c illustrates the electronic band structure of the monolayer g-SiC3. The g-SiC3 is a zero bandgap semiconductors, in which the lowest conduction band and the highest valence band meet at the Fermi level at the high symmetry K point. The junction of the conduction and valence bands is equivalent to the Dirac point of graphene. Moreover, we calculated the partial density of states (PDOS) to study the nature of electron hybridization in the system. As shown in Fig. 1d, the lowest conduction band are mainly contributed form C atoms, while the C and Si atoms give comparable contributions to the highest valence band.
3.2 Electronic properties of bilayer g-SiC3
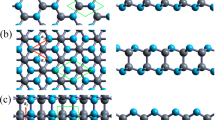
It is well known that the interaction between layers play a decisive role in the properties of layered materials. Thus, changing the stacking model could provide us a simple but effective way to modify the electronic properties of layered bilayer g-SiC3. Based on the results of monolayer g-SiC3, we studied the electronic properties of the bilayer g-SiC3. As shown in Fig. 2, here, three different stacking models are considered: (1) AA stacking, in which all C (or Si) atoms in upper SiC3 layer lie above the C (or Si) atoms in lower SiC3 layer; (2) AB is Bernal stacking, where the monolayer g-SiC3 is rotated by 60° around the Si atom and then stacked with another g-SiC3 layer; (3) AC stacking, in which the monolayer g-SiC3 is rotated by 60° around the C atom and then stacked with another g-SiC3 layer. These three stacking types can be mutual transformed through translational sliding and rotation of the basal plane. The optimized structure parameters of bilayer g-SiC3 with different stacking models are shown in Table 2. The binding energies (Eb) have been calculated to determine the most stable bilayer structure. The binding energy of bilayer system is defined as

Top and side views of optimized structures (upper panels), deformation charge density (middle panels) and electronic band structures (lower panels) of bilayer g-SiC3 with different stacking models. The blue dashed lines and orange solid lines represent the depletion and accumulation of electron, respectively. The dashed black line is the Fermi energy level (set as 0)
where \({E}_{g-SiC3}\) and \({E}_{Bilayer}\) are the total energy of the monolayer and bilayer g-SiC3 system, respectively. Among all three stacking models, the AA stacking bilayer g-SiC3 with smallest binding energy is most stable.
As we can see from Table 2, for all bilayer systems considered, the lattice constants ‘a’ are almost the same (5.75 Å for AA stacking model, 5.74 Å for AB and AC stacking models). Therefore, the bond lengths, i.e., dC-C and dC-Si are also around the same values. However, the interlayer spacings (d) of bilayer g-SiC3 systems exhibit relatively large deviations. The layer spacing of the AA and AB stacked system are almost the same (2.426 Å and 2.488 Å), while the interlayer spacing of the AC is 3.579 Å. By contrast, the bond length of Si–Si in monocrystalline silicon is 2.328 Å ~ 2.384 Å [30–32], which is very close to the value of Si–Si bond length in the AA and AB stacking bilayer g-SiC3 systems. In addition, in AA stacking bilayer g-SiC3 systems, we found the distance between two C (or Si) atoms is 3.259 Å (or 2.426 Å). They are in good agreement with the previously reported values [17] (3.33 Å between two carbon atoms and 2.41 Å between two silicon atoms). These results suggest that our calculation methods are reasonable for the bilayer g-SiC3 system. Therefore, there is a strong interaction between the Si atoms in upper SiC3 layer and Si atoms in lower SiC3 layer. On the other hand, the AC stacking bilayer g-SiC3 is stabilized mainly through Van der Waals interaction. As shown in Fig. 2d–f, for all three stacking methods, the Si–C bonds within each SiC3 layer is covalent, which is similar to the nature of a single layer [21]. Moreover, between two SiC3 layers, the Si–Si bonds in AA and AB stacking bilayer g-SiC3 are strongly covalent, while no obvious interaction was found between the g-SiC3 layer in AC stacking model. All Si atoms in the AA stacking model participated in the covalent bonding, while in the AB stacking model, the covalent bonding can only be found in some part of the intermediate regions between two g-SiC3 layers. This also reasonably explains the longer interlayer spacing in AA stacking bilayer g-SiC3 system. This can also explain why the amplitude of buckling (Δ) of the AA and AB stacking models is larger than that in the AC stacking model.
To discuss the electronic properties of different stacking models, we also considered the energy band structures of bilayer g-SiC3. It can be seen from Fig. 2g–i that the AA stacking bilayer g-SiC3 is an indirect semiconductor, the CBM and VBM are located the high symmetry Г and M points. The contributions from C and Si atoms to the band structures of AA stacking bilayer g-SiC3 system are presented in Fig. 3a and b. Near the Fermi level, the CBM is mainly contributed from C atoms, while the VBM is contributed from C and Si atoms. The bandgap calculated by PBE and HSE06 are 1.38 eV and 1.959 eV, respectively. However, the AB and AC stacking bilayer g-SiC3 are shown to be metallic. Since the AA stacking is most stable, in the following study we mainly focused on the tunable band structure of AA stacking bilayer g-SiC3 system.

Projected band structures of (a) C atoms and (b) Si atoms in AA stacking bilayer g-SiC3. (c) The total and partial density of states of AA stacking bilayer g-SiC3
3.3 Electronic properties of bilayer AA stacking g-SiC3 under biaxial and vertical stress
Recent experiments show that stretching strain is an effective method to modify the mechanical properties composites [33, 34]. To develop its promising application in the field of optoelectronics, we also investigated the electronic properties of bilayer g-SiC3 systems under different biaxial stress. Since the AA stacking bilayer g-SiC3 is the most stable and the only semiconductor among all the three stacking models, we take a further discussion on the electronic structures of AA stacking bilayer g-SiC3 under different compression and stretching strains. The tension/compression ratio of the bilayer structures is expressed by the following formula:
where \(a_{{0}}\) and \(a\) represents the lattice constant of the unstrained and strained bilayer g-SiC3, respectively. Here, \(\Delta a\)(\(a{ - }a_{{0}}\)) represents the change in the lattice constant after the application of the biaxial strain. To further study the effect of strain engineering on the energy band structures of AA-SiC3 bilayer systems, we calculated the contribution of the Si and C atoms of bilayer systems with different biaxial strain. In Fig. 4, the blue and orange lines represent the projected band structure produced by Si atoms and C atoms, respectively. As shown in Fig. 4, when the compressive strain changes from 0% to − 3%, the VBM moves from M point to the position between M and Γ point, while the CBM moves from Γ point to M point. Interestingly, when the tensile stress range from 1 to 3%, the bilayer g-SiC3 system change from indirect semiconductor to a zero bandgap semimetal. When the tensile stress reaches 3%, near the K point, there are two Dirac points at the Fermi level. This band structures are similar to that of the bilayer graphene. As shown in the projected band structures, when the tensile stress reaches 3%, since the electronic states around Fermi level near K point are all contributed from carbon atoms, the unique band structures for the bilayer g-SiC3 are caused by the interactions of C atom layers only. What’s more, there is small buckling in the SiC3 layer of unstrained bilayer AA g-SiC3 system. However, the strained bilayer AA g-SiC3 with tensile stress of 3% became a planar structure, which promotes the sp2 hybridization between C and Si atoms.

Projected band structures of (upper panels) C atoms and (lower panels) Si atoms in AA stacking bilayer with different biaxial strains
In addition, experiments show that the bandgap structures can also be affected by the interlayer spacing between two adjacent layers. Thus, we further investigated the bandgap values of the AA g-SiC3 bilayer systems under various vertical strain. In this article, the vertical strain is define as:
where \(d_{0}\) and \(d\) denote the equilibrium and strained interlayer distance of SiC3 bilayer systems, respectively. It can be seen from Fig. 5 that, as the interlayer distance is increased, all bands move toward the deeper energy level. Due to the increase of interlayer interaction, the bandgap decreases with the interlayer spacing increasing. Moreover, when the interlayer spacing is larger than its equilibrium value, as the interlayer spacing increasing, the CBM moves from Γ point to M point. When the tensile stress applied on the SiC3 bilayer reaches d = 2.83 Å (∆d = 0.4 Å), the CBM and VBM both located at the M point, which suggests the transformation from an indirect bandgap semiconductor to a direct bandgap semiconductor through vertical stretching. These results offer a convenient way to control the bandgaps of SiC3 bilayer systems.

Energy band structures of AA-stacked g-SiC3 under vertical strain
4 Conclusion
We have performed a systematical study of structural and electronic properties of bilayer g-SiC3. Based on first-principle calculation, the monolayer g-SiC3 is a zero bandgap semiconductor. For the three bilayer g-SiC3 systems, the most stable structure is the AA stacking model, which is an indirect bandgap semiconductor. The bandgap calculated by PBE and HSE06 are 1.380 eV and 1.959 eV, respectively. Moreover, for the most stable AA stacking bilayer g-SiC3, the bandgap values and types of could be modulated by applying biaxial or vertical compressing/stretching. The bilayer g-SiC3 could turn into a zero bandgap semiconductor with the in-plane biaxial stretching strain reaches 3%. On the other hand, the bilayer g-SiC3 could also turn into a direct bandgap semiconductor with the vertical tensile stress reaches d = 2.83 Å (∆d = 0.4 Å). The tunable band structures make g-SiC3 a promising candidate for future nanodevices.
References
N.O. Weiss, H. Zhou, L. Liao, Y. Liu, S. Jiang, Y. Huang, X. Duan, Graphene: An emerging electronic material. Adv. Mater. 24, 5782–5825 (2012). https://doi.org/10.1002/adma.201201482
T. Mueller, F. Xia, P. Avouris, Graphene photodetectors for high-speed optical communications. Nat. Photonics 4, 297–301 (2010). https://doi.org/10.1038/nphoton.2010.40
R. Summary, 2D materials and van der Waals heterostructures. Appl. Phys. (2016). https://doi.org/10.1126/science.aac9439
J.M. Raimond, M. Brune, Q. Computation, F. De Martini, C. Monroe, Electric Field Effect in Atomically Thin Carbon Films 306, 666–670 (2004). https://doi.org/10.1126/science.1102896
K.S. Novoselov, D. Jiang, F. Schedin, T.J. Booth, V.V. Khotkevich, S.V. Morozov, A.K. Geim, Two-Dimens. Atom. Crys. 102, 10451–10453 (2005). https://doi.org/10.1073/pnas.0502848102
S. Zhang, X. Xu, T. Lin, P. He, Recent advances in nano-materials for packaging of electronic devices. J. Mater. Sci.: Mater. Electron. 30, 13855–13868 (2019). https://doi.org/10.1007/s10854-019-01790-3
M. Yang, S.W. Kim, S. Zhang, D.Y. Park, C.W. Lee, Y.H. Ko, H. Yang, Y. Xiao, G. Chen, M. Li, Facile and highly efficient fabrication of robust Ag nanowire-elastomer composite electrodes with tailored electrical properties. J. Mater. Chem. C. 6, 7207–7218 (2018). https://doi.org/10.1039/c8tc01691g
G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. Chen, M. Chhowalla, Coherent atomic and electronic heterostructures of single-layer MoS2. ACS Nano 6, 7311–7317 (2012). https://doi.org/10.1021/nn302422x
Y.Y. Hui, X.F. Liu, W.J. Jie, N.Y. Chan, J.H. Hao, Y.T. Hsu, L.J. Li, W.L. Guo, S.P. Lau, Exceptional Tunability of Band Energy in a Compressively Strained Trilayer MoS2 Sheet. ACS Nano (2013). https://doi.org/10.1021/nn4024834
X.D. Li, S. Yu, S.Q. Wu, Y.H. Wen, S. Zhou, Z.Z. Zhu, Structural and electronic properties of superlattice composed of graphene and monolayer MoS2. J. Phys. Chem. C 117, 15347–15353 (2013). https://doi.org/10.1021/jp404080z
S. Cahangirov, M. Topsakal, E. Aktürk, H. Šahin, S. Ciraci, Two- and one-dimensional honeycomb structures of silicon and germanium. Phys. Rev. Lett. 102, 1–4 (2009). https://doi.org/10.1103/PhysRevLett.102.236804
B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, Ś Vizzini, B. Ealet, B. Aufray, Epitaxial growth of a silicene sheet. Appl. Phys. Lett. 97, 223107–223109 (2010). https://doi.org/10.1063/1.3524215
A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang, Y. Yamada-Takamura, Experimental evidence for epitaxial silicene on diboride thin films. Phys. Rev. Lett. 108, 1–5 (2012). https://doi.org/10.1103/PhysRevLett.108.245501
M. Tahir, P. Vasilopoulos, Electrically tunable magnetoplasmons in a monolayer of silicene or germanene. J. Phys.: Condens. Matter 27, 75303 (2015). https://doi.org/10.1088/0953-8984/27/7/075303
P. Vogt, P. Capiod, M. Berthe, A. Resta, P. De Padova, T. Bruhn, G. Le Lay, B. Grandidier, Synthesis and electrical conductivity of multilayer silicone. Appl. Phys. Lett. 104(2), 021602 (2014). https://doi.org/10.1063/1.4861857
G. Gao, N.W. Ashcroft, R. Hoffmann, The unusual and the expected in the Si/C phase diagram. J. Am. Chem. Soc. 135, 11651–11656 (2013). https://doi.org/10.1021/ja405359a
T. Hussain, A.H. Farokh Niaei, D.J. Searles, M. Hankel, Three-Dimensional Silicon Carbide from Siligraphene as a High Capacity Lithium Ion Battery Anode Material. J. Phys. Chem. C (2019). https://doi.org/10.1021/acs.jpcc.9b06151
F. Zheng, H. Dong, Y. Ji, Y. Li, Adsorption and catalytic decomposition of hydrazine on metal-free SiC3 siligraphene. Appl. Surf. Sci. 469, 316–324 (2019). https://doi.org/10.1016/j.apsusc.2018.11.002
M. Houmad, M.H. Mohammed, R. Masrour, A. El Kenz, A. Benyoussef, Electronic and electrical properties of siligraphene (g-SiC3) in the presence of several strains. J. Phys. Chem. Solids 127, 231–237 (2019). https://doi.org/10.1016/j.jpcs.2018.12.016
X. Qin, Y. Wu, Y. Liu, B. Chi, X. Li, Y. Wang, X. Zhao, Origins of Dirac cone formation in AB3 and A3B (A, B = C, Si, and Ge) binary monolayers. Scientific Reports. 7, 1–13 (2017). https://doi.org/10.1038/s41598-017-10670-x
Y. Ding, Y. Wang, Geometric and electronic structures of two-dimensional SiC3 compound. J. Phys. Chem. C 118, 4509–4515 (2014). https://doi.org/10.1021/jp412633y
M. Zhao, R. Zhang, Two-dimensional topological insulators with binary honeycomb lattices: SiC3 siligraphene and its analogs. Physical Review B - Condensed Matter and Materials Physics. 89, 1–8 (2014). https://doi.org/10.1103/PhysRevB.89.195427
C. Pereyra Huelmo, P.A. Denis, Silicon carbide induced doping of graphene a new potential synthetic route for SiC3 siligraphene. J. Phys. Chem. C 123, 30341–30350 (2019). https://doi.org/10.1021/acs.jpcc.9b07978
P.E. Blöchl, Projector augmented-wave method. Physical Review B. 50, 17953–17979 (1994). https://doi.org/10.1103/PhysRevB.50.17953
G. Kresse, J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B-Condensed Matter and Materials Physics. 54, 11169–11186 (1996). https://doi.org/10.1103/PhysRevB.54.11169
J.P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). https://doi.org/10.1103/PhysRevLett.77.3865
G. Kresse, J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metalamorphous- semiconductor transition in germanium. Physical Review B. 49, 14251–14269 (1994). https://doi.org/10.1103/PhysRevB.49.14251
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Journal of Chemical Physics (2010). https://doi.org/10.1063/1.3382344
M.S. Dresselhaus, A. Jorio, R. Saito, Characterizing graphene, graphite, and carbon nanotubes by Raman spectroscopy. Annual Review of Condensed Matter Physics. 1, 89–108 (2010). https://doi.org/10.1146/annurev-conmatphys-070909-103919
X.K. Lu, T.Y. Xin, Q. Zhang, Q. Xu, T.H. Wei, Y.X. Wang, Versatile mechanical properties of novel g-SiC x monolayers from graphene to silicene A first-principles study. Nanotechnology (2018). https://doi.org/10.1088/1361-6528/aac337
J. Heyd, J.E. Peralta, G.E. Scuseria, R.L. Martin, Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional. Journal of Chemical Physics 10.(1063/1), 2085170 (2005)
L. Stauffer, J.L. Bischoff, L. Kubler, D. Aubel, L. Simon, G. Garreau, C. Pirri, P. Sonnet, M. Stoffel, A. Selloni, A. De Vita, R. Car, Atomic structure of carbon-induced (formula presented) reconstruction as a Si-Si homodimer and C-Si heterodimer network. Physical Review B-Condensed Matter and Materials Physics. 64, 1–9 (2001). https://doi.org/10.1103/PhysRevB.64.035306
J. Kim, J. Kim, S. Song, S. Zhang, J. Cha, K. Kim, H. Yoon, Y. Jung, K.W. Paik, S. Jeon, Strength dependence of epoxy composites on the average filler size of non-oxidized graphene flake. Carbon 113, 379–386 (2017). https://doi.org/10.1016/j.carbon.2016.11.023
M.L. Jin, S. Park, J.S. Kim, S.H. Kwon, S. Zhang, M.S. Yoo, S. Jang, H.J. Koh, S.Y. Cho, S.Y. Kim, C.W. Ahn, K. Cho, S.G. Lee, D.H. Kim, H.T. Jung, An Ultrastable Ionic Chemiresistor Skin with an Intrinsically Stretchable Polymer Electrolyte. Adv. Mater. 30, 1–9 (2018). https://doi.org/10.1002/adma.201706851
Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities, China under Grant No. 2412019FZ037.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Niu, R., Li, X., Guan, Y. et al. Electronic properties of bilayer g-SiC3 system. J Mater Sci: Mater Electron 32, 1888–1896 (2021). https://doi.org/10.1007/s10854-020-04957-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-04957-5