
Abstract
Non-stoichiometric ZnS1−x:Mnx nanomaterials were prepared using a thermolysis procedure by decreasing the stoichiometric amount of thiourea relative to the amount of zinc acetate as starting precursors: Zn(Ac)/(1−x) thiourea in the presence of (x) manganese acetate (x = 0, 0.01, 0.03, 0.05, and 0.1). Rietveld X-ray diffraction analysis was used to examine the structural modification in the ZnS lattice induced by sulfur deficiency and Mn2+ incorporation. The samples with x ≤ 0.01 exhibited a single ZnS zincblende phase, while other samples, x ≥ 0.02, have two phases ZnS and ZnO with different percentages. The lattice parameter of the system is governed by the Mn and S amounts in the matrix. A high-resolution transmission electron microscope established the quantum dot nature of the system. Fourier-transform infrared technique confirmed the presence of ZnS and ZnO phases in higher Mn-doped ZnS samples. The bandgap obtained from UV–vis analysis showed non-monotonic dependence on Mn content; it initially decreased and then increased to form a bandgap “bowing.” Photoluminescence analysis revealed that the emission colors depended on the amount of Mn doping in the matrix. The PL intensity raised for all Mn-doped samples as compared with the pristine sample reached its maximum value for the 3% and 5% Mn samples. The PL exhibited a red shift for the high dopant amount of Mn. Density function theory calculation was used to explore the electronic and optical characteristics of pure phases in ZnS1−xMnx system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Luminescent nanosemiconductors’ compounds have attracted great attention recently because of their unusual physical and chemical characteristics [1,2,3,4,5,6,7,8,9,10]. Among II–VI semiconductors materials, zinc sulfide (ZnS) has a large direct bandgap of 3.6–3.9 eV, a small exciton Bohr radius of 2.5 nm, a high refractive index (n = 2.57), and a large exciton binding energy (40 meV) [11,12,13]. Therefore, it can be used in different applications such as light-emitting diodes, solar cells, infrared windows, bio-labels, lasers, display tubes, and sensors [14]. Moreover, ZnS formed in two crystallographic forms: hexagonal and cubic phases [15].
On the other hand, nano-ZnS material is more suitable in a large range of applications rather than its bulk counterpart due to the following reasons [16,17,18]: (i) fast recombination of electron–hole pairs generated in bulk ZnS limits its application in photocatalysis, (ii) the limited excitation wavelength of the bulk ZnS (less than 340 nm), (iii) nano-ZnS displays quantum confinement effect with a significant blue shift in absorption spectra, (iv) larger surface-to-volume ratio, and (v) nano-ZnS has long luminescence lifetime.
Furthermore, the electrical conductivity, magnetic and luminescent characteristics of semiconductors materials can be modified by doping it with different transitional metals such as Mn, Co, and Fe [19]. Such kinds of doping can nominate this material to be used in other different promising applications such as light-emitting devices, bio-labeling, biological imaging, and spin-based electronics [20,21,22,23]. Additionally, a number of studies indicate that cubic ZnS is a much superior host for doping and photoluminescence [24]. For example, doping ZnS with Mn caused a reduction of the self-absorption and boosting quantum efficiency [25]. Furthermore, the insertion of Mn2+, which has 3d5 configurations [26] in ZnS matrix, leads to interaction between the d electrons of Mn and s-p electrons of ZnS [27]. This interaction makes Mn2+ plays as a potential emission center [28], therefore, the relaxation of electrons from the excited state to the ground state of incorporated Mn ion in the ZnS lattice caused emission of the orange color [28]. Mn-doped ZnO(1-x)Sx nanoparticles exhibited a strong orange emission and its bandgap energies increased as Mn-doped amount increased in the matrix [29]. It was found that the S2− to Zn2+ molar ratio in Mn-doped ZnS quantum dot controlled the Mn2+ 4T1(G)–6A1(S) emission. As the molar ratios of S2− to Zn2 increased from 0.5:1 to 3.5:1, the PL intensity of orange-red-to-blue color ratio enhanced [30].
Additionally, more significant modifications in the electronic and optical characteristics can be performed by partial substitution of the anions of different materials by another anions or cations [31,32,33]. For example, the absorption of ZnO or TiO2 in the visible region was enhanced by substituting its oxygen ions with nitrogen [31]. Furthermore, substituted sulfur ions by P and Cl ions in hexagonal CdS and ZnS structures help in enhancement of hydrogen evolution [32]. Mohamed et al. demonstrated that the bandgaps of CdS1−xMnx thin films can be reduced from 2.437 to 2.230 eV, but the linear and non-linear refractive index were enhanced as 10% sulfur ions of CdS were substituted by Mn [33].
There are many methods used to synthesize sulfide materials, but the thermolysis method used in this work is simple, low price, mass scale amount process, no solvent to be used in the reaction and consequently no waste disposal issues associated with the solvent are required to be taken into account.
The aim of the present study is to investigate influence of the Zn/S ratio and Mn doping on the structural and optical properties of the obtained samples. In the present work, nano-ZnS1−x:Mnx (x = 0, 0.01, 0.03, 0.05, and 0.1) was synthesized using thermolysis method. The effect of increasing of Zn and Mn amounts as compared with the sulfur amount in the matrix on the structural, microstructural, optical and electronic characteristics of the formed samples was performed in details by X-ray diffraction, High-resolution transmission electron microscope (HRTEM), Fourier-transform infrared (FTIR) spectrometer, UV–Vis, Photoluminescence techniques and Castep software based on density function theory calculation (DFT).
2 Experimental details
Nano-ZnS1−x:Mnx (x = 0.0, 0.01, 0.03, 0.05, and 0.1) samples were prepared using a thermolysis procedure by decreasing the stoichiometric amount of thiourea (SC(NH2)2, 99%, Sigma-Aldrich) relative to the amount of zinc acetate dihydrate (Zn(CH3COO)2.2H2O, 98%, Sigma-Aldrich) as starting precursors: Zn(Ac)/(1−x) thiourea in the presence of (x) Manganese acetate tetrahydrate (Mn(CH3COO)2· 4H2O, 98%, Sigma-Aldrich) (x = 0, 0.01, 0.03, 0.05, and 0.1); heated at 300 °C (3 h) in an electric oven in air [34,35,36,37,38,39,40], see the preparation schema in Fig. 1. X-ray diffraction data were collected by X’pert MPD, Philips (Cu-Kα, 2θ = 10°–80°, step = 002°, counting time = 2 s). The X-ray diffraction data were analyzed applying Rietveld method [41] and using MAUD software [42] to obtain the lattice parameter, crystallite size, occupancies of different cations and percentage of the phases developed in different samples. The instrumental broadening was corrected using LaB6 standard. A high-resolution transmission electron microscope (HRTEM) was used to examine the morphology and nanosize scale of the studied samples. UV–Vis measurements were obtained using UV-3101PC UV–Vis–NIR spectrophotometer; for photoluminescence (PL) Shimadzu RF1501 spectrofluorometer with a resolution of 10 nm was used, and Bruker Tensor 27 FTIR spectrometer for Fourier-transform infrared (FTIR) spectra. Density functional calculation (DFT) was performed for using Cambridge sequential total energy package [43] (CASTEP) code based on first principle calculation. The following criteria was applied during the calculation: generalized gradient approximation [44] (GGA) for the exchange–correlation function in the framework of Perdew–Burke–Ernzerhof (PBE) [45], ultrasoft pseudopotentials, supercell of ZnS (cubic structure) with dimension 2 × 1 × 1, energy cutoff (350 eV), maximal force between the atoms (0.01 eV/Å) and the self-consistent (5 × 10− 6 eV/atom). In the case of ZnS/Mn, one sulfur atom was substituted with a sulfur atom and the above criteria were applied. The 3d54s2, 3d104s1 and 3s23p4 electrons relevant to Zn, Mn and S atoms were employed for the pseudopotential configurations, respectively.

Scheme of preparation for nano-ZnS1−xMnx system
3 Results and discussion
3.1 XRD structural analysis
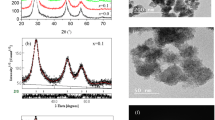
To investigate the structural modification in the ZnS lattice induced by sulfur deficiency and Mn2+ incorporation, Rietveld XRD analysis is performed applying the MAUD program [41, 42]. All the XRD patterns of ZnS1−xMnx samples, Fig. 2a, exhibited broad peaks typical of quantum dot structure. For Mn content x ≤ 0.01, XRD patterns corresponding to pure ZnS of cubic zincblende structure, maybe there is ZnO phase with a small percentage in the sample and it cannot be detected by XRD. For x ≥ 0.03, besides the major zincblende, another phase is formed which identified to be hexagonal ZnO. Accordingly, Mn ions should be incorporated into the ZnS lattice either substitutionally for Zn ions or interstitially at voids available in the cubic zincblende structure, namely the tetrahedral holes (¾ ¾ ¾) or octahedral holes at (½ ½ ½). Meantime, S deficiency allows some O ions to be incorporated into the lattice for compensation. Rietveld structural analysis was performed to determine the crystallographic positions and the occupancies of Mn and O ions incorporated into the ZnS lattice. The structural parameters obtained from Rietveld analysis are listed in Table 1 and the XRD fitting resulting for x = 0.0 and 0.1 are depicted in Fig. 2b, c. Results manifested that O atoms are incorporated into the ZnS lattice substitutionally for S atoms at 4c (¼¼¼) positions. Bad reliability factors for Rietveld refinement were obtained when trying to accommodate O atoms at the interstitial voids. Similar incorporation of O atoms into chalcogenides was found upon annealing ZnTe [46] and ZnSe [47] at different temperatures. To determine how Cu is accommodated, the occupancy of Cu ions is set free to refine in the last cycle of Rietveld refinement assuming Mn replacing Zn at (000) and residing interstitially at (¾ ¾ ¾) or (½ ½ ½); keeping the total occupancy equal to the value of (x). The resulting occupancy for the interstitial sites was almost zero indicating that Cu is incorporated in the lattice mainly substitutionally for Zn. The average crystallite size for all samples is in the range of quantum dots and this is confirmed from HRTEM image together with SAED pattern for ZnS0.99Mn0.01, where the particles are agglomerated and have a very small in size (less than 5 nm), Fig. 2d, e. As shown in Table 1, the lattice parameter (a) decreased upon increasing the Mn content (sulfur deficiency) up to x = 0.03 and increased for x ≥ 0.05. The lattice parameter (a) is affected by many factors; Mn (0.66 Å) doping increased its value, while S deficiency decreased it. Also, the Mn occupancy at the Zn position (000) is less than the nominal value indicating that some Mn ions are incorporated interstitially. For Mn content x ≥ 0.02, part of Zn ions segregated and form the ZnO phase which increased with (x) reaching 5% for x = 0.1.

a X-ray diffraction patterns for ZnS1−xMnx, b, c Rietveld refinement for ZnS1-xMnx with x = 0.0, 0.1, and d, e TEM image and SAED for nano-ZnS0.99Mn0.01 sample
3.2 Infrared absorption spectroscopy analysis
FTIR spectra of ZnS1−xMnx with x = 0, 0.01, 0.03, 0.05, and 0.1 are revealed in Fig. 3. The zincblende unit cell belongs to Td point group and contains four atoms [48] and according to group theory, it has A1, A2, E, T1 and 2T2 phonon modes. Only T2 mode is active in both Raman and IR spectroscopy [48]. The characteristic major bands in ZnS1−xMnx system are observed in the ranges 432–445, 602–613 and 1022–1049 cm−1 which are corresponding to asymmetric bending, symmetric bending, and asymmetric stretching, respectively [49]. The absorption peaks near 1551 and 1401 cm−1 are attributable to carboxyl group band and CH2 bending, respectively [50,51,52]. Small bands observed from 400 to 600 cm−1 are referred to ZnS matrix [53], which originates from the resonance interaction between vibrational modes of sulfide ions in the crystal. The characteristic vibration of the Zn–O stretching bond around 430 cm−1 [54,55,56] overlaps with that of ZnS 426–444 cm−1. Furthermore, the IR bands of ZnS1−x:Mnx are shifted toward larger wavenumber (Table 2); this can be explained based on the XRD analysis result, where Mn is incorporated in the lattice substitutionally for Zn (Mn has less mass than Zn), therefore the band shifted to larger wavenumber.

FTIR spectra of ZnS1-xMnx nanosystem
3.3 UV absorption analysis
To clarify the effect of incorporation Mn content (x) instead of S on the optical characteristics of ZnS, the UV–vis absorption spectra are measured for the samples with x = 0, 0.01, 0.02, 0.03, 0.05, and 0.1 in the wavelength range of 300–1100 nm, Fig. 4. Since ZnS has a direct bandgap transition, the optical bandgap (Eg) was determined by applying the Tauc equation [57] in the form:
where hν, α and A are the photon energy, absorption coefficient and a constant, respectively. The bandgap energy (Eg) for each sample was determined from the (αhυ)2-hν relation by extrapolating the linear part and find the intersection with the abscissa axis, hν. The obtained values of bandgap energies are given in Table 3. For the pure ZnS sample, the bandgap energy is 3.93 eV which is large compared to that 3.6 eV of bulk material [58]; this increase of the bandgap can be ascribed to the quantum confinement effect of the ZnS nanosize. For doped samples, two bandgaps are determined corresponding to two prominent slopes representing two distinct absorption onsets representing the existence of two different phases ZnS and ZnO. The bandgaps show non-monotonic dependence on Mn content; it initially decreased, down to 2.89 eV for manganese content x = 0.03, and then increased for x = 0.05 and 0.1, although the size of the nanocrystals is unaffected with altering Mn-doped amount. It is expected the bandgap to decrease with raising Mn content, as the bandgap of MnS (Eg ~ 3.1 eV) is smaller than that of ZnS (Eg ~ 3.6 eV) and ZnO (Eg ~ 3.4 eV). However, the increase of Eg at higher concentrations of Mn needs explanation. Such non-monotonic change of the bandgap is found in Mn-doped ZnO nanocrystals [59] and in Mn-doped CdS [60] where an initial reduction in the bandgap occurs, after an enhancement upon increasing the dopant; the energy bandgaps minimum position relies on the particle size. However, the opposite trend has been observed in Cu-doped ZnO nanocrystal [61] and in Mn-doped ZnS [62]; the bandgap exhibited first a reduction and then an enhancement with the increasing amount of dopant. Such initial anomalous change in the bandgap is attributed to a bandgap “bowing” in a very short-ranged, paramagnetic phase of Mn-doped: ZnO [59], CdS [60], and ZnS [62]. The second-order perturbation theory is used to explain the band bowing in the Mn-doped ZnSe [63]; it was ascribed to the strong exchange interactions present between the d electrons of Mn and the s and p electrons of the host bands. Similarly, the non-monotonic change of the bandgap in the present ZnMnxS1−xO2x may be attributed to the bandgap “bowing” resulting from these short-range exchange interactions. Also, such bandgap bowing is observed for ZnS1−Ox and is explained by first principles calculations [64]. For low values of oxygen (x), the conduction band minimum (CBM) is shifted to lower energies but the valence band remaining largely fixed, which means a reduction in the bandgap energy; for high values, the conduction band remaining largely fixed and the valance band maximum (VBM) shifted to lower energies, so the bandgap energy increases again (Table 3).

Plot of (ahυ)2 vs hυ for ZnS1−xMnx samples
3.4 Photoluminescence analysis
Figure 5a–e reveals the Gaussian fitting for PL spectra of the undoped and Mn-doped ZnS nanocrystals under excitation wavelength 325 nm. An obvious three sub-emission peaks at 365 (UV), 412(violet) and 417(violet) nm are detected for ZnS, Table 3. The dopant can play a significant task in the host matrix, which can successfully change the material’s microstructure and characteristics. Samples doped with 1 and 3% Mn (ZnS0.99Mn0.01 and ZnS0.97Mn0.03) showed also three sub-peaks at (361(UV), 379(UV), 404(violet) nm) and (361(UV), 378(UV), 401(UV) nm), respectively. On the other hand, samples with 5 and 10% Mn (ZnS0.95Mn0.05 and ZnS0.9Mn0.1) revealed four and five sub-peaks (359 (UV), 376(UV), 383(violet), 454(blue) nm) and (370(UV), 381(blue), 431(blue), 456(blue), 523 (green) nm), respectively, Table 3. Many authors [65,66,67] showed that nano-ZnS prepared by different methods has a blue emission band, while Zn1-xMnxS samples have blue and orange bands. Curcio et al. [68] revealed that ZnS prepared by the solvothermal method has four blue-green emissions. On the other hand, Manimegalai et al. [69] established that Mn-doped ZnS (Mn = 30 and 50%) synthesized by a facile microwave combustion method emitted blue color only.

a–e Gaussian fitting for photoluminescence spectrum and (f) the PL peak position and PL maximum intensity for ZnS1-xMnx samples under 325 nm
It is well known that the existence of point defects (interstices and vacancies) in a semiconductor material created new energy levels (defect states) inside the forbidden bandgap. In the case of ZnS, zinc interstices and vacancies of sulfur operated as donors (electron traps), while sulfur interstices and vacancies of zinc worked as acceptors (hole traps). Many radiative electron transitions could occur between the defect states and the conduction band (or the valence band), caused a broad and defect-state emissions [70].
The UV and violet emissions are due to dangling orbitals of sulfur residing on the surface and the recombination of carriers between the sulfur vacancies related donor and the valence band [70], respectively. Furthermore, the blue band emission and the tail of the PL spectrum, extending beyond 500 nm, are ascribed to the existence of intrinsic defects in ZnS like sulfur vacancies on the nanocrystal surface or zinc vacancies inside the structure [70]. Additionally, the green color is the character feature for Zn2+ as luminescent centers in the lattice [68].
On the other hand, the increase of either the amount of Zn2+ or S2− ions in the reaction solution can create defect states inside the forbidden bandgap of the ZnS host [71]. These defect states lead to quenching the intensity of Mn2+: 4T1(G)–6A1(S) emission; this is because of the radiative recombination happening at states rather than energy transfer to Mn2+ energy levels [68].
Nguyen et al. [72] found that as the amount of sulfide is low as compared with the amount of zinc in the ZnS: Mn2+ QDs matrix, a large number of sulfur vacancies (VS) states are produced inside the lattice structure which is working as donor centers (electron traps). Consequently, the orange-red emission intensity originating from Mn2+ ions is less than the blue emission intensity of the ZnS host. The result pointed out that the transfer energy from the ZnS host to Mn2+ energy levels is not effective owing to surface states.
The PL intensity of the present study is raised for all Mn-doped samples as compared with undoped ZnS, reaching its maximum value for the sample with 3% and 5% Mn (ZnS0.97Mn0.03 and ZnS0.95Mn0.05), Fig. 5f. It is concluding that the incorporation of Mn into the ZnS matrix reduced the defects and made it a better matrix as luminescent materials. A similar effect was noticed in Mn-doped ZnS (Zn1−xMnxS) nanocrystals were synthesized using co-precipitation procedure [69]. Also, it can be noticed that the PL exhibited a redshift at higher values of Mn incorporated into ZnS matrix, Fig. 5f. A similar effect was observed in ZnS(1%)Mn/ZnO core–shell nanostructures, where the PL emissions shifted toward the red region due to the increase of ZnO amount in the matrix.
3.5 Electronic properties
The calculated lattice constants are a = 5.2759(5) and 5.3109(6) Ǻ for ZnS and ZnS/Mn obtained from GGA method. These results are in close agreement with XRD result; the lattice parameter of ZnS was increased as it doped with Mn.
The spin polarized electronic band structures of the ZnS and ZnS0.9Mn0.1 samples were calculated based on their optimized lattice constants within the GGA scheme. Figure 6 reveals the electronic band structures of both samples along with the directions of high symmetry in the first Brillouin zone. One can notice the semiconductor nature with a conduction band minimum (CBM) and a valence band maximum (VBM) at the G point for ZnS with direct energy gap 2.28 eV, which is lower than the experiment value of 3.53 eV, see the UV part. The bandgap underestimation of DFT always occurs because of the well-known limitation of estimating accurate energy band properties [38]. The energy gap of ZnS decreased as part of S in ZnS sample substituted with Mn, but still the ZnS/Mn sample exhibited a direct transition. When the transition metal (Mn) substitutes sulfur in ZnS lattice, a strong p-d exchange interaction occurred between Mn-3d and anion S-3p orbital and caused a metallic nature of the material.

a, b Electronic band structure for ZnS and ZnS0.9Mn0.1 samples
Additionally, the electronic characteristics (total and partial density of states) of undoped and doped ZnS with Mn reveal in Figs. 7, 8. From the total density of state (TDOS), it is noticed that the valence band formed from two parts in both samples. The upper part width of the valence band is 7.1 eV for ZnS and it increased to 8.9 eV as ZnS doped with Mn. Furthermore, the valence band maxima (VBM) are basically formed from the p states of sulfur and zinc atoms close to the Fermi level in the case of ZnS, see partial density of state (PDOS). But the lower part of the upper part of valence band (VB) is basically formed from S-p and Zn-d states. Additionally, the bottom of the conduction band is mainly created from S–s states and a little part out from s, p, d states of the Zn atom. For ZnS/Mn, the VBM shifted to the lower energy and it created from S-p, Zn-p and Mn-d. The lower part of the upper part of VB of the valence band is formed mainly from S-p and Zn-d. The major contribution to the bottom of the conduction comes from S–s, s, p, d states of the Zn atom. In addition, the conduction band for ZnS sample is formed from one part divided into two regions. ZnS doped with Mn behaves a similar trend but it shifted toward Fermi level, therefore the energy gap of Mn-doped ZnS sample decreased. The conduction band is consistent mainly from (i) S-p and s and p- states of Zn atom in case of ZnS and (ii) S-p, Zn- s and p and Mn-p states in case of ZnS/Mn sample.

Total density of states (TDOS) for ZnS and ZnS0.9Mn0.1 samples

Partial density of states (PDOS) for ZnS and ZnS0.9Mn0.1 samples
The different optical parameters such as absorption coefficient, α(ω) and reflectivity, R(ω) can be estimated using the real (ε1) and imaginary (ε2) parts of dielectric functions (ε(ω) = ε1(ω) + iε2(ω)) as indicated in the following equations [30]:
where ω, p and k are the angular frequency, the main part of the Cauchy integral and extinction coefficient, respectively.
Figure 9a reveals the absorption coefficient of the samples as function of the electromagnetic wave energy. This parameter described the decay of light intensity per unit distance in the material. The absorption edge is located at > 3.5 eV with maximum values at 6.52 and 11.05 eV, for the sample with x = 0.0. The ZnS/Mn sample exhibited the maximum absorption edge at 7.43 and 11.54 eV and extra peaks at 0.61 and 2.89 eV as compared with ZnS. This new peaks may be associated with the present of Mn ions in the matrix, as revealed in Fig. 7.

Optical functions a absorption and b, c dielectric function for ZnS and ZnS0.9Mn0.1 samples
Nevertheless, the absorption coefficient rose quickly for lower energy, which is ascribed to the fact that more transitions between valence and conduction bands may happen in these energy ranges. Such quick enhancement in the absorption coefficient above the absorption edge is characteristic of semiconductors materials [73]. Comparing the absorption diagrams of the undoped and Mn-doped ZnS samples, it is noticed that the doped sample exhibited a higher absorption at IR frequencies region.
Figure 9b, c shows the ε1 and ε2 parts of the dielectric function for ZnS and ZnS/Mn samples. The static dielectric constant, which is known as the zero-frequency limit of ε1(0) [74], was enhanced with substitution of some S atoms with Mn atoms in ZnS. This indicates that a reduction in the energy gap of ZnS/Mn as compared with ZnS has been occurred. Figure 9b reveals also the dielectric peaks (ε1) of ZnS/Mn sample are shifted to lower energy as compared with ZnS sample. This shift indicates great energy absorption.
It has been noticed for undoped ZnS that ε1 (ω) attained a maximum value at 4.2 eV and it reached a minimum value at 6.8 eV. As some S atoms in ZnS matrix substituted with Mn atoms, ε1(ω) has a maximum value at 1.8 eV. Then, it reduced and became negative reaching a minimum around of 7.3 eV. The negative value pointed out that the incident light wave is completely reflected in this region and the material displays a metallic behavior. Furthermore, the imaginary part of the dielectric function is related to the absorption of electromagnetic waves because of the inter-band transitions. The imaginary part of the dielectric function, Fig. 9, showed an extra peak in the low energy part for ZnS/Mn as compared with undoped ZnS, which can be ascribed to the difference in electronic structure between the undoped and doped systems.
4 Conclusion
XRD analysis together with a high-resolution transmission electron microscope (HRTEM) established the quantum dot nature of ZnS1−xMnx system. The lattice parameter decreased upon increasing the Mn content (sulfur deficiency) up to x = 0.03 and increased for x ≥ 0.05. Occupancy analysis of different cations in the matrix indicated that some Mn ions incorporated interstitially in the ZnS matrix. ZnO phase is formed due to the segregation of Zn ions in the matrix. The characteristic major bands in ZnS and ZnO were confirmed by FTIR technique. The bandgap initially decreased for manganese content x = 0.03, and then increased for x = 0.05 and 0.1. PL analysis showed that ZnS and 1% Mn doping emitted UV and violet colors while the sample with 3% Mn doping exhibited UV only. On the other hand, samples with higher Mn concentration revealed UV, violet and blue colors but sample with 10% Mn emitted green color in addition. DFT explained the decrease of the energy gap as ZnS doped with Mn. The doped sample exhibited a higher absorption in the IR range. All the previously mentioned properties nominate the ZnS1-xMnx system to be employed in many applications such as LEDs, visible light-photocatalytic activity and solar energy applications.
References
Z.K. Heiba, M.B. Mohamed, Opt. Quant. Electron. 52(2), 99 (2020)
M.M.S. Sanad, M.M. Rashad, J. Mater. Sci. Mater. Electron. 27, 9034 (2016)
M.M.S. Sanad, M.M. Rashad, E.A. Abdel-Aal, M.F. El-Shahat, K. Powers, J. Electron. Mater. 43(9), 3559 (2014)
M.S. Eraky, M.M.S. Sanad, E.M. El-Sayed, A.Y. Shenouda, E.S. El-Sherefy, AIP Adv. 9(11), 115115 (2019)
Z.K. Heiba, M.B. Mohamed, N.Y. Mostafa, A.M. El-Naggar, Inorg Organomet Polym 30, 1898 (2020)
Z.K. Heiba, M.B. Mohamed, N.Y. Mostafa, Int. J. Appl. Ceram. Technol. 16(4), 1590 (2019)
Z.K. Heiba, M.B. Mohamed, N.G. Imam, J. Supercond. Novel Magn. 32(9), 298 (2019)
Z.K. Heiba, M.B. Mohamed, Appl. Phys. A 124(12), 818 (2018)
Z.K. Heiba, M.B. Mohamed, A.M. Wahba, N.G. Imam, J. Electron. Mater. 47(1), 711 (2018)
Z.K. Heiba, N.G. Imam, M.B. Mohamed, J. Mol. Struct. 1115, 156 (2016)
X.S. Fang, T.Y. Zhai, U.K. Gautam, L. Li, L.M. Wu, Y. Bando, D. Golberg, Prog. Mater. Sci. 56, 175 (2011)
R.M. Krsmanović Whiffena, D.J. Jovanović, Ž. Antić, B. Bártová, D. Milivojević, M.D. Dramićanin, M.G. Brik, J. Lumin. 146 (2014) 133.
A.I. Cadis, L.E. Muresan, I. Perhaita, V. Munteanu, Y. Karabulut, J.G. Guinea, A. Canimoglu, M. Ayvacikli, N. Can, Opt. Mater. 72, 533 (2017)
W. Xua, X. Meng, W. Ji, P. Jing, J. Zheng, X. Liu, J. Zhao, H. Li, Chem. Phys. Lett. 532, 72 (2012)
M.B. Mohamed, Int. J. Appl. Ceram. Technol. (2019). https://doi.org/10.1111/ijac.13336
A.L. Stroyuk, A.E. Raevskaya, A.V. Korzhak, S.Y. Kuchmii, J. Nanopart. Res. 9, 1027 (2007)
D. Chen, F. Huang, G. Ren, D. Li, M. Zheng, Y. Wang, Z. Lin, Nanoscale 2, 2062 (2010)
H. Yang, S. Santra, P.H. Holloway, J. Nanosci. Nanotechnol. 5, 1364 (2005)
Z.K. Heiba, M.B. Mohamed, J Inorg Organomet Polym 30(3), 879 (2020)
M.Y. Chen, C.C. Chang, Jpn. J. Appl. Phys. 48, 112201 (2009)
M. Labrenz, G.K. Druschel, T. Thomsen-Ebert, B. Gilbert, S.A. Welch, K.M. Kemner, G.A. Logan, R.E. Summons, G.D. Stasio, P.L. Bond, B. Lai, S.D. Kelly, J.F. Banfield, Science 290, 1744 (2000)
H. Pang, Y. Yuan, Y. Zhou, J. Lian, L. Cao, J. Zhang, X. Zhou, J. Lumin. 122, 587 (2007)
J. Cai, S. Wang, K. Zhu, Y. Wu, L. Zhou, Y. Zhang, Q. Wu, Xi. Wang, Z. Hu, RSC Adv. 8 (2018)374.
A. Nag, S. Chakraborty, D.D. Sarma, J. Am. Chem. Soc. 130, 10605 (2008)
S.R. Chalana, V.S. Kavitha, R. Reshmi Krishnan, V.P.M. Pillai, J. Alloys Compounds 771 (2019) 721
G. Murugadoss, V. Ramasamy, M.R. Kumar, Appl. Nanosci. 4, 449 (2014)
R.N. Bhargava, D. Gallagher, Phys. Rev. Lett. 72, 416 (1994)
H. Yang, J. Zhao, L. Song, L. Shen, Z. Wang, L. Wang, D. Zhang, Mater. Lett. 57, 2287 (2003)
F.B. Dejene, M.O. Onani, L.F. Koao, A.H. Wako, S.V. Motloung, M.T. Yihunie, PhysicaB 480, 63 (2016)
T.P. Nguyen, Q.V. Lam, T.B. Vu, J. Lumin. 196, 359 (2018)
A. Roy, C.N.R. Raoa, APL Materials 7, 090901 (2019)
S. Kouser, S.R. Lingampalli, P. Chithaiah, A. Roy, S. Saha, U.V. Waghmare, C.N.R. Rao, Angew. Chem. 127, 8267 (2015)
M. Mohamed, A.M. Abdelraheem, M.I. Abd-Elrahman, N.M.A. Hadia, E.R. Shaaban, Appl. Phys. A 125, 483 (2019)
Z.K. Heiba, M.B. Mohamed, N.G. Imam, J. Mater. Sci.: Mater. Electron. 26(9), 6344 (2015)
M.B. Mohamed, Z.K. Heiba, N.G. Imam, J. Mol. Struct. 1163, 442 (2018)
Z.K. Heiba, M.B. Mohamed, Appl. Phys. A 124(6), 446 (2018)
Z.K. Heiba, M.B. Mohamed, M.H.A. Kader, J. Electron. Mater. 47(5), 2945 (2018)
Z.K. Heiba, M.B. Mohamed, N.Y. Mostafa, Appl. Phys. A 125(2), 132 (2019)
Z.K. Heiba, M.B. Mohamed, N.G. Imam, Int. Polym. Proc. 33(2), 226 (2018)
Z.K. Heiba, A.A. Albassam, M.B. Mohamed, Appl. Phys. A 126, 479 (2020)
L. Lutterotti, Nucl. Inst. Methods, Phys. Res. B. 268, 334 (2010).
J. Rodríguez-Carvajal, Phys. B (Amsterdam, Neth.) 192, 55 (1993).
J. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson, M.R. Pederson, D.J. Singh, C. Fiolhais, Phys. Rev. B Condens. Matter 46, 6671 (1992)
J.P. Perdew, Y. Wang, Phys. Rev. B 45, 13244 (1992)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett 77, 3865 (1996)
Z.K. Heiba, Cryst. Res. Technol. 38(6), 488 (2003)
Z.K. Heiba, Powder Diffr. 17(3), 191 (2002)
E. Kroumova, M. I. Aroyo, J. M. Perez Mato, A. Kirov, C. Capillas, S. Ivantchev, H. Wondratschek, Phase Transitions 76(1–2), 155 (2003).
J.X. Yang, S.M. Wang, X. Zhao, Y.P. Tian, S.Y. Zhang, B.K. Jin, X.P. Hao, X.Y. Xu, X.T. Tao, M.H. Jiang, J. Crystal Growth 310, 4358 (2008)
R. Devi, P. Purkayastha, P.K. Kalita, B.K. Sarma, Bull. Mater. Sci. 30(2), 123 (2007)
R. Seoudi, A.B. El- Bailly, W. Eisa, A.A. Shabaka, S.I. Soliman, R.K. Abd El Hamid, R.A. Ramadan, J. Appl. Sci. Res. 8(2), 658 (2012).
S.B. Qadri, E.F. Skelton, D. Hsu, A.D. Dinsmore, J. Yang, H.F. Gray et al., Phys Rev B. 60, 9191 (1999)
B. S. Rema Devi, R. Raveendran, A. V. Vaidyan. Pramana-J. Phys. 68, 679 (2007).
P.G. Devi, A.S. Velu, J. Mater. Sci.: Mater. Electr. 27, 10833 (2016)
A. Sadollahkhani, I. Kazeminezhad, J. Lu, O. Nur, L.H.M. Willander, RSC Adv. 4, 36940 (2014)
J. Huso, L. Bergman, M.D. McCluskey, J. Appl. Phys. 125, 075704 (2019)
J. Tauc, R. Grigorovici, A. Vancu, Phys. Status Solidi 15, 627 (1966)
M. Jaquez, K. M. Yu, M. Ting, M. Hettick, J. F. S_anchez-Royo, M. Wełna, A. Javey, O. D. Dubon, W. Walukiewicz, J. Appl. Phys. 118, 215702 (2015).
R. Viswanatha, S. Sapra, S.S. Gupta, B. SatpatiP, V. Satyam, B.N. Dev, D.D. Sarma, J. Phys. Chem. B 108, 6303 (2004)
L. Levy, J.F. Hochepied, M.P. Pileni, J. Phys. Chem. 100, 18322 (1996)
R. Viswanatha, S. Chakraborty, S. Basu, D.D. Sarma, J. Phys. Chem. B 110(45), 22310 (2006)
S. Sapra, J. Nanda, A. Anand, S.V. Bhat, D.D. Sarma, J. Nanosci. Nanotechnol. 3, 392 (2003)
R.B. Bylsma, W.M. Becker, J. Kossut, U. Debska, D. Yoder-Short, Phys. ReV. B 33, 8207 (1986)
C. Persson, C. Platzer-Björkman, J. Malmström, T. Törndahl, M. Edoff, Phys. Rev. Lett. 97, 146403 (2006)
S. Sapra, A. Prakash, A. Ghangrekar, N. Periasamy, D.D. Sarma, J. Phys. Chem. B 109, 1663 (2005)
R. M. K. Whiffen, D. J. Jovanović, Ž. Antić, B. Bártov, D. Milivojević, M. D. Dramićanin, M. G. Brik, J. Luminescence 146, 133 (2014).
T.T.Q. Hoa, N.D. The, S.M. Vitie, N.H. Nam, L.V. Vu, T.D. Canh, N.N. Long, Opt. Mater. 33(3), 308 (2011)
A.L. Curcio, L.F. da Silva, M.I.B. Bernardi, E. Longo, A. Mesquita, J. Lumin 206, 292 (2019)
A. Manikandan, D. K. Manimegalai, S. Moortheswaran, D. S. Arul Antony, J Supercond Nov Magn. 28(9), 2755 (2015).
N.T. Tuan, D.Q. Trung, N.V. Quang, N.D. Hung, N.T. Khoi, P.T. Huy, P.F. Smet, K.W. Meert, D. Poelman, J. Lumin. 199, 39 (2018)
R. Viswanath, H.S. Bhojya Naika, G.S. Yashavanth Kumar, P.N. Prashanth Kumar, K.N. Harisha, M.C. Prabhakara, R. Praveen, Appl. Surf. Sci. 301, 126 (2014).
R. Hempelmann 52. J.K. Salem, T. M. Hammad, S. Kuhn, I. Nahal, M. A.Draaz, N. K. Hejazy, J. Mater. Sci.: Mater. Electron. 25, 5188 (2014).
S.Z. Karazhanov, P. Ravindran, A. Kjekshus, H. Fjellvag, B.G. Svensson, Phys. Rev. B 75, 155104 (2007)
Z.K. Heiba, M.B. Mohamed, N.M. Farag, A.M. El-naggar, A.A. Albassam, J. Mater. Sci.: Mater. Electron (2020). https://doi.org/10.1007/s10854-020-03821-w
Acknowledgements
The authors are grateful to the Deanship of Scientific Research, King Saud University for funding through Vice Deanship of Scientific Research Chairs.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Heiba, Z.K., Mohamed, M.B., Ahmed, S.I. et al. Structural, optical, and electronic properties of non-stoichiometric nano-ZnS1−x: Mnx. J Mater Sci: Mater Electron 31, 13447–13459 (2020). https://doi.org/10.1007/s10854-020-03898-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-03898-3