
Abstract
Hf4+ substituted M-type hexaferrites with composition of BaHfxFe12−xO19 (x = 0–0.08 in step of 0.02 ) were synthesized via a solid reaction method. Pure M phase was observed by XRD analysis excepted when x = 0.08, the second phase (HfO2) appeared when x = 0.08. With increase of the Hf4+ substitution contents, the average grain size decreased, the shape of grains became irregular gradually and some small particles appeared and spacing between grains increased. It could be explained by lattice distortion and the agglomeration of grains. In terms of magnetic properties, the saturation magnetization (Ms) decreased from 81.16 emu/g to 38.36 emu/g and the coercivity (Hc) increased from 666 Oe to 1220 Oe as x increased from 0.00 to 0.08, which might be attributed to occupancies of Hf4+ ions on 2b sublattice. This would result in a valence change of Fe3+ to Fe2+ at the 2a site. Moreover, the magnetocrystalline anisotropy of the samples maintained a high level and changed slightly, while the saturation magnetization of the samples decreased to a low level, which provided a choice for magnetic materials to meet higher frequency applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
M-type barium hexaferrite (BaM), with composition of BaFe12O19, has relatively high saturation magnetization, large coercivity, large magnetic crystalline anisotropy, excellent chemical stability, and high Curie temperature [1,2,3,4,5,6,7]. Since been discovered in 1957, BaM has been widely used not only in permanent magnets but also in microwave and millimeter devices (such as circulator, isolator) [3, 8,9,10]. To meet higher frequency application, it is of great impotence to keep the magnetocrystalline anisotropy of barium ferrite at a high level and change the saturation magnetization of materials to a low level simultaneously. Fortunately, it is an advantage of hexaferrite that the saturation magnetization can be adjusted through substitution for Fe3+.
Ion substitution has attracted much attention due to easy operability and high efficiency. So far, there are two main approaches to ion substitution that have been reported. One is the single ion substitution for Fe3+ ions [11,12,13]. The advantage of this way is the introduction of variable uniqueness and easy control. But the limitation is that only trivalent ions can be introduced for the substitution of Fe3+ ions to maintain charge balance. The other way is double ions substitution: simultaneously using one divalent ion and one tetravalent ion to replace two Fe3+ ions [14, 15]. In this study, we choose Hf4+ to substitute one Fe3+ ion. Hf belongs to transition metals. The ionic radius of Hf4+ is 0.71 Å, which is slightly bigger than that of the Fe3+ ion (0.645 Å), so it is possible for Hf4+ ions to occupy Fe3+ ions sites. The electronegativity of Hf (1.32) is smaller compared with that of Fe (1.83). The doping of Hf4+ will lead to the change of lattice structure. Furthermore, it will also lead to the change of Fe3+ into Fe2+, which will affect the magnetic properties of M-type barium ferrite. Many researchers have reported double ions substitution simultaneously using one divalent ion and one tetravalent ion to replace two Fe3+ ions. But a few of reports concentrate on single tetravalent ion substitution. In this paper, we got the single Hf4+ substitution BaM via a solid reaction method, then investigated the effect of low-level substitutions of Hf4+ on the microstructure and magnetic properties of BaM.
2 Experiment procedure
Hf4+ substitution of M-type barium ferrite (BaHfxFe12−xO19, x = 0.00, 0.02, 0.04, 0.06, and 0.08) were synthesized using analytical grade BaCO3, Fe2O3, HfO2 via solid-state reaction. All of the raw materials were in powder form, first all of the powders were mixed using ball milling for 12 h in a small Teflon canikin with zirconia balls and deionized water as the milling media. Next the mixed powders was dried and pre-sintered at 1100 ℃ for 4 h in air. Then, the pre-sintered powders were milled again for 12 h under the same condition as the first mill. After drying, add moderate 8 wt% of polyvinyl alcohol (PVA) as a binder to make the granule and press it into 2–3-mm-thick plates and ring. Finally, the compacted samples were sintered at 1300 ℃ for 15 h.
The phase compositions and the lattice constant of the samples were determined using an X-ray diffractometer (XRD.Empyrean.PANalyticalB.V.).The bonding situation and component was analyzed by XPS test (Thermo Scientific K-Alpha+).The microstructures of the samples were characterized using a scanning electron microscope (SEM.FEG 250.Quanta). Magnetization hysteresis loops were measured using a vibrating sample magnetometer (VSM.PPMS.Quantum Design).
3 Result and discussion
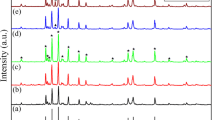
The XRD patterns of BaHfxFe12−xO19 (x = 0.00, 0.02, 0.04, 0.06) ferrites sintered at 1300 °C with different x contents are shown in Fig. 1. With the increase of x content, all the samples exhibited the typical peaks of the pure hexagonal ferrite phase. All diffraction peaks were indexed to the M-type barium hexaferrite phase (space group P63/mmc, JCPDS File Number 43-0002) with low substitution content of Hf4+ (x = 0.00, 0.02, 0.04, 0.06), which shows that Hf4+ successfully enters the internal structure of M-type barium ferrite. However, when the content of Hf4+ increased to 0.08, the second phase (HfO2) appeared in the XRD patterns. We conjectured that some HfO2 is located at the grain boundaries without entering the internal structure. It indicated that the doping limit of Hf4+ for M-type barium ferrite in this process is less than 0.08.

XRD patterns of BaHfxFe12−xO19 with different x values
The lattice constants a and c can be calculated based on XRD data and Scherrer’s equation described as following:
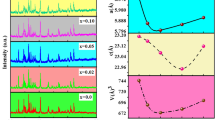
Among them, d is interplanar distance and h, k, and l are indices of crystal face [16]. The variations of lattice constants a and c keep decreasing with increase of Hf4+ substitution content as shown in Fig. 2. It could be explained in two respects. One is the influence of the Hf4+ ionic radius, which is bigger than the Fe3+ ion, the doping of Hf4+ would increase the lattice constants. The other respect is the formation of Fe2+. According to the conservation of valence states, doping Hf4+ would inevitably lead to the formation of Fe2+ and the existence of Fe2+ would cause lattice distortion, which affected the growth of lattice and resulted to the decrease of the lattice constants in turn. Combining the above two respects, lattice constant showed a downward trend as shown in Fig. 2.

Lattice constant with different Hf4+ substitution contents

XPS spectra for Fe with different Hf4+ substitution contents

XPS spectra for Hf with different Hf4+ substitution contents
Fe2p XPS spectra of different Hf4+ substitution contents are showed in Fig. 3. The signal of the high-binding energy tail with five 3d electron (3d5) was strong, and the main peeks appeared at about 710 ev (Fe2p3/2) and 724 ev (Fe2p1/2). The shake-up appeared in main peeks between all samples. With the increase of x content, only the peeks moves in the direction of lower-binding energy (the peak of Fe2+) in turn without other significantly changes. It means Fe2+ and Fe3+ co-exist in all samples, but with the increase of Hf4+ content, some Fe3+ gradually changes to Fe2+ so that the electrical valence could be balanced, just as shown in Fig. 3.
The XPS spectra for Hf of all samples are showed in Fig. 4. All the samples exhibited the similar peaks. With the increase of x content, the peak intensity increased significantly in two positions (Hf4f5/2 and Hf4f7/2), and these two positions are the split peaks of the Hf–O bonds, just as shown in Fig. 4. It could be explained as following: in this whole experiment, Hf is only bonding with O, so with the increase of Hf4+ content, the amount of Hf–O bonds increases. In combination with Fig. 3, with the increase of Hf4+ content, the amount of Hf–O bonds increased, and some Fe3+ gradually changes to Fe2+ so that the electrical valence could be balanced.

SEM micrographs of sintered BaHfxFe12−xO19 with different Hf4+ substitution contents
The micrographs of sample powders were characterized by SEM and are shown in Fig. 5. Some typical hexagonal grains are shown in Fig. 5a. The calculated average grain size was about 3 µm. With the increase of Hf4+ substitution contents, the average grain size decreased. The shape of grains became irregular gradually. Some small particles appeared. The spacing between grains increased. The porosity of sample increased and the compactness degradated. This could be explained that doping Hf4+ would inevitably lead to the formation of Fe2+ and thus would cause lattice distortion and the agglomeration of grains.

M–H hysteresis loops of the samples with different Hf4+ substitution

Coercivity and saturation magnetization with different Hf4+ substitution contents
The Magnetic hysteresis loop measurements of samples are shown in Fig. 6. Figure 7 shows values of saturation magnetization (Ms) and coercivity (Hc) of samples. Ms decreased from 81.16 to 38.36 emu/g and Hc increased from 666 Oe to 1220 Oe with x increased from 0.00 to 0.08. It is known that Fe3+ ions are respectively in 5 different lattice position 2a (↑), 4f2 (↓), 12k (↑) (octahedral), 2b (↑) hexahedron, and 4f1 (↓) tetrahedral. The magnetic moments of Fe3+ ions at 2a (↑), 2b (↑), and 12 k (↑) positions are parallel to each other due to the superexchange between Fe ion and O ion, while the magnetic moments of Fe3+ ions at 4f1 (↓) and 4f2 (↓) lattices are inversely parallel to the magnetic moments at the above three locations [17]. The net magnetic moments determine the saturation magnetization. When Hf4+ ions occupied these sites instead of Fe3+ ions, the superexchange interaction would change, which leaded to the change of Ms in turn. From the Ligand occupancy theory, it can be seen that for M-type barium ferrite, the ion occupancy usually depends on the number of electrons in the outer d-orbit of the ion. When the number of electrons in the d-orbit is 1–4, the doped ions will preferentially replace the Fe3+ in the tetrahedral position. When the number of electrons in the d-orbit is 6–9, the doped ions will preferentially replace the Fe3+ in the octahedral position, and when the number of electrons in the d-layer orbitals is 0, 5, and 10, there is no selectivity [18]. Therefore, there are no preferential sites for Hf4+ ions because of their d0 configuration. In general, the electronegativity of ions is larger. The lattice position of the octahedron with larger gap space should be replaced first, then the hexahedron and then the tetrahedron [19]. The electronegativity of Fe3+ and Hf4+ are 1.83 and 1.32, respectively. Hence, the Hf4+ might prefer to replace the position of the hexahedron (2b↑) or tetrahedral (4f1↓). In addition, the ionic radius of Hf4+ is bigger than that of the Fe3+ ion, and due to the volume effect, the possibility of Hf4+ entering tetrahedron is small. So the Hf4+ is most likely to replace the position of the hexahedron (2b↑) and then the Fe3+ at 2a position would change to Fe2+ to maintain the valence equilibrium [20]. The result of saturation magnetization is shown in Fig. 7. It might be caused by the substitution of Hf4+ for Fe3+. On the one hand, the Bohr magnetic moment of Hf4+ is smaller than that of Fe3+, so the total magnetic moment decreased which leaded to the decrease of the saturation magnetization. On the other hand, the substitution of Hf4+ changed the equilibrium of Fe2+ and Fe3+ at the 2a site and reduced the superexchange interaction between Fe ion and O ion in hexaferrite system which also leaded to the decrease of the saturation magnetization. It is also shown in Fig. 7 that in the beginning of doping (x ≤ 0.06) the saturation magnetization (Ms) drops rapidly. However, with the increase of Hf4+ doping amount (0.06 ≤ x ≤ 0.08), the decreasing rate of Ms decreased. It could be attributed to the appearance of HfO2 phase, which means some HfO2 was located at the grain boundaries rather than occupying the position of Fe3+; these part of Hf4+ had no contribution to the net magnetic moments. Moreover, HfO2 is a non-magnetic phase, which just dilutes the magnetic properties of the whole material. So it would slow down the downward trend of Ms. The change of saturation magnetization showed that the substitution of Fe3+ ions by Hf4+ ions is an effective way to modify the saturation magnetization of materials.
It is known that coercivity (Hc) is largely influenced by magnetocrystalline anisotropy. To clarify the changing mechanism of Hc, we further calculated the magnetocrystalline anisotropy constant (K1) and the magnetocrystalline anisotropy field (Hk) as shown in Table 1.
Hf4+ ion replaced Fe3+ ion and caused the increase of Fe2+ ion content. Hf4+ ion occupied more Fe3+ ion positions. The valence state transition of Fe3+ ion leads to the change of magnetic moment orientation ratio and magnetocrystalline anisotropy. The magnetocrystalline anisotropy constant (K1) decreased with the increase of Hf4+ doping amount. It might reduce the coercivity to some extent. However, doping of Hf4+ ion could also lead to lattice distortion of crystal structure, reduced particle size and increased grain boundaries of the crystals, so that there were more locations to hinder the movement and rotation of the magnetic domains in turn lead to the increase of coercivity. Finally, under their combined action, the coercivity had a relatively small increase with the increase of Hf4+ doping amount.
The values of coercivity (Hc) versus Hf4+ substitution contents are given in Fig. 7. As Stoner–Wohlfarth theory described, the coercivity (Hc) of ferrite can be determined by the saturation magnetization (Ms) and the magnetocrystalline anisotropy constant (K1) as following equation [21]:
K1 is magnetocrystalline anisotropy constant; the saturation magnetization (Ms) can be determined using the law of near saturation.
In high H field region, Mh exhibited a linear tendency as a function of H2 since parameters a and χp became small enough to be ignored, which provide an approach to the value of parameter b and Ms [22].
Once parameter b was identified, K1 could also be obtained as following Eq. (4):
The result of K1 as shown in Table 1 decreased sharply from 2.96 × 105 to 1.2 × 105 J/m3 with increasing of x from 0 to 0.08. It has been reported that the Fe3+ ions at 2b and 4f2 sites make remarkable contribution to the magnetocrystalline anisotropy. For a single Fe3+ ion at 2b, 4f2, 2a, 4f1, and 12 k site, its contribution to the magnetocrystalline anisotropy is 1.4, 0.51, 0.23, 0.18, − 0.18, respectively [23]. Only Fe3+ ions at 12k site have negative consequences on magnetocrystalline anisotropy. Therefore, when Hf4+ ions occupied 2b sites and which the Fe3+ turned to Fe2+ at 2a position. They all resulted to the decrease of magnetocrystalline anisotropy constant in turn leading to the sharp reduction of K1 as shown in Table 1.
Moreover, the magnetocrystalline anisotropy field (Hk) was also calculated according to the following Eq. (5):
where µ0 is the universal constant of permeability in free space, 4π*10− 7 H/m [1]. It is precisely because K1 and Ms decrease rapidly at the same time that lead to a little decrease of Hk just as seen in Table 1.
4 Conclusion
BaHfxFe12−xO19 (0 ≤ x ≤ 0.08) samples were successfully synthesized by a sintered temperature at 1300 °C. When 0 ≤ x ≤ 0.06, only one crystal phase existed in the sample, and when x = 0.08, there were HfO2 phase appearance. With the increase of x content, the Fe2p XPS spectra peeks moves to low-binding energy, The Hf4f XPS spectra peak intensity increased significantly in Hf4f5/2 and Hf4f7/2. The morphology of the grains were shown to be gradually irregular. The lattice parameters and the saturation magnetization were sharply decreased, the magnetocrystalline anisotropy field had a slight decrease and the values of coercivity was increased with the increase of the Hf4+ substitution. The result could be explained by the substitution of Hf4+ and the change of their structure, ion occupancy, and micro-morphology. Moreover, the result also provides a choice for magnetic materials to meet higher frequency application.
References
R.C. Pullar, Hexagonal ferrites: a review of the synthesis, properties and applications of hexaferrite ceramics. Prog. Mater. Sci. 57, 1191–1334 (2012)
H.M. Khan, M.U. Islam, Y.B. Xu, M.A. Iqbal, I. Ali, Structural and magnetic properties of TbZn-substituted calcium barium M-type nano-structured hexaferrites. J. Alloys Compd. 589, 258–262 (2014)
V.G. Harris, A. Geiler, Y. Chen, S.D. Yoon, M. Wu, A. Yang, Z. Chen, P. He, P.V. Parimi, X. Zuo, Recent advances in processing and applications of microwave ferrites. J. Magn. Magn. Mater. 321, 2035–2047 (2009)
Y. Wan, Y. Li, Y. Guo, Q. Zheng, X. Wu, C. Xu et al., Structure, ferroelectric, piezoelectric and ferromagnetic properties of BiFeO3-Ba0.6(Bi0.5K0.5)0.4TiO3 lead-free multiferroic ceramics. J. Mater. Sci. Mater. Electron. 25, 1534–1541 (2014)
C.K. Ong, H.C. Fang, Z. Yang, Y. Li, Magnetic relaxation in Zn-Sn-doped barium ferrite nano-particles for recording. J. Magn. Magn. Mater. 213(3), 413–417 (2000)
Y.S. Yuan, Tuo, Microwave adsorption of Sr(MnTi)(x)Fe12–2xO19 particles. J. Magn. Magn. Mater. 342, 47–53 (2013)
V.G. Harris, Modern microwave ferrites. IEEE Trans. Magn. 48, 1075–1104 (2012)
F.Y. Guo, G.J. Ji, J.J. Xu, H.F. Zou, S.C. Gan, X.C. Xu, Influence of Tb substitution on electromagnetic and microwave absorption properties of barium hexaferrites. Mater. Res. Innov. 18, 112–119 (2014)
C.J. Li, B. Wang, J.N. Wang, Magnetic and microwave absorbing properties of electrospun Ba(1–x)LaxFe12O19 nanofibers. J. Magn. Magn. Mater. 324, 1305–1311 (2012)
H. Sozeri, I. Kucuk, H. Ozkan, Improvement in magnetic properties of La substituted BaFe12O19 particles prepared with an unusually low Fe/Ba molar ratio. J. Magn. Magn. Mater. 323, 1799–1804 (2011)
D.M. Chen, Y.L. Liu, Y.X. Li, K. Yang, H.W. Zhang, Microstructure and magnetic properties of Al-doped barium ferrite with sodium citrate as chelate agent. J. Magn. Magn. Mater. 337, 65–69 (2013)
S.M. El-Sayed, T.M. Meaz, M.A. Amer, H.A. El Shersaby, Magnetic behavior and dielectric properties of aluminum substituted M-type barium hexaferrite. Phys. B Condens. Matter 426(1), 137–143 (2013)
C. Singh, S.B. Narang, I.S. Hudiara, Y. Bai, F. Tabatabaei, Static magnetic properties of Co and Ru substituted Ba-Sr ferrite. Mater. Res. Bull. 43, 176–184 (2008)
H. Sozeri, H. Deligoz, H. Kavas, A. Baykal, Magnetic, dielectric and microwave properties of M-Ti substituted barium hexaferrites (M = Mn2+, Co2+, Cu2+, Ni2+, Zn2+). Ceram. Int. 40, 8645–8657 (2014)
W.J. Zhang, Y. Bai, X. Han, L. Wang, X.F. Lu, L.J. Qiao, Magnetic properties of Co-Ti substituted barium hexaferrite. J. Alloys Compd. 546, 234–238 (2013)
J. Li, H. Zhang, Y. Liu, Q. Li, G. Ma, H. Yang, The transformation behavior of Mtype barium ferrites due to Co-Ti substitution. J. Mater. Sci.: Mater. Electron. 26, 4668–4674 (2015)
D. Chen, Y. Liu, Y. Li, W. Zhong, H. Zhang, Microstructure and magnetic properties of low-temperature sintered CoTi-substituted barium ferrite for LTCC application. J. Magn. Magn. Mater. 323, 2837–2840 (2011)
Y. Qian Liu, C. Liu, Wu Yu, J. Wang, L. Li, H. Gao Zhang, Investigation on Zn-Sn co-substituted M-type hexaferrite for microwave applications. J. Magn. Magn. Mater. 444, 421–425 (2017)
M. Jazirehpour, M. Shams, O. Khani, Modified sol-gel synthesis of nanosized magnesium titanium substituted barium hexaferrite and investigation of the effect of high substitution levels on the magnet properties. J. Alloy. Compd. 545, 32–40 (2012)
J. Li, H. Zhang, Y. Li, Q. Li, G. Yu, Effect of La-Zn Substitution on the structure and magnetic properties of low temperature co-fired m-type barium ferrite. J. Supercond. Nov. Magn. 27, 793–797 (2014)
Z. Zi, Q. Liu, J. Dai, Y. Sun, Effects of Ce-Co substitution on the magnetic properties of M-type barium hexaferrites. Solid State Commun. 2012, 894–897 (2012)
J.H. You, H.H. Kim, I. Yoo, Preparation of strontium W-type hexaferrites in a low oxygen pressure and their magnetic properties. J. Alloy. Compd. 695, 3011–3017 (2017)
Z. Yang, C. Wang, X. Li, H. Zeng, (Zn, Ni, Ti) substituted barium ferrite particles with improved temperature coefficient of coercivity. Mater. Sci. Eng. B 90, 142–145 (2002)
Funding
This work was supported by the National Natural Science Foundation of China [Grant Numbers 51872041].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, L., Liu, Y., Liu, Q. et al. Effect of single Hf4+ ion substitution on microstructure and magnetic properties of hexagonal M-type Ba(Hf)xFe12−xO19 ferrites. J Mater Sci: Mater Electron 31, 4106–4112 (2020). https://doi.org/10.1007/s10854-020-02957-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-02957-z