
Abstract
Herein, reduced graphene oxide (RGO) and magnetic reduced graphene (RGO–MNP) nanosheets were synthesized by using Fe2+ ions via the facile and green method for the first time. Prepared nanomaterials were characterized by UV–Vis, FTIR, Raman, XRD, VSM and TEM techniques. Interaction of RGO, GO, RGO–MNP and GO–MNP nanosheets with two valance metal ions were investigated by tracing square wave voltammetry of a modified carbon paste electrode (CPE) responses. Both modified CPE by RGO–MNP and GO–MNP showed higher response between ions to Pb2+ ion. In optimized experimental and instrumental conditions, a linear calibration curve from 1.0 × 10−9 to 1.0 × 10−3 M Pb2+ with detection limit as 3.07 × 10−9 M Pb2+ was observed for modified CPE by GO–MNP, and two linear calibration curve from 1.0 × 10−9 to 5.0 × 10−6 and from 1.0 × 10−5 to 1.0 × 10−3 M Pb2+ with detection limit as 8.13 × 10−10 M Pb2+ were observed for modified CPE by RGO–MNP. Prepared sensors showed good stability, sensitivity, repeatability and reproducibility in this work.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Graphene with a two-dimensional structure has high stability and its interesting properties like, elastic, thermal and electrical led to great consideration in recent years [1]. Today, varieties of methods are used to synthesize graphene. The most common of which are mechanical methods, including micro mechanic peeling of graphite [2], dry ice [3] and chemical methods, including chemical vapor deposition [4] and reduction of graphene oxide [5]. By these methods, graphene can be made with a fairly complete structure and excellent properties.
Following the research on graphene, the graphite oxide discovered about 150 years ago and production of it was first reported in 1840 by Schafhaeutl [5] and in 1859 by Brodie [6]. The proposed method has two important characteristics: (a) Graphite is used as a raw material that is economically feasible [7] and can provide a high percentage of graphene in large scale. (b) The resulting graphene oxide is hydrophilic [8] and can form a stable aqueous colloid solution [9]. Besides, the reduction of graphene oxide will partially repair the structure and properties of graphene [10]. Different methods of reducing graphene oxide create different properties of graphene, which can affect its final performance. Therefore, graphene oxide and its reduction are key issues in the research on the production of graphene [11,12,13].
The chemical structure of graphene oxide was first tested by Dreyer et al. [14]. Recently, the methods proposed by Hummers and Offeman in 1958 [15] are often used to produce graphene oxide in which the graphite is oxidized to graphite oxide using sulfuric acid, sodium nitrate and potassium permanganate. The reduction of graphene oxide can cause a large change in its structure and properties such as apparent features [16], electrical conductivity, and carbon to oxygen ratio [17]. Some of which are important and should be considered in the process of resuscitation. Reduction of graphene oxide involves thermal [18], radiation [19, 20], chemical [16, 21,22,23,24], photocatalysis [25, 26], electrochemical [27, 28], solvothermal [29,30,31], and multi-step [32,33,34,35] reduction methods.
In general, the reduction of graphene oxide is accomplished to achieve two goals: elimination of functional groups [3, 36] and the repair of structural defects [37]. The mechanisms of graphene oxide reduction can be divided into three general categories; (a) removal of oxygenating functional groups from the surface by mentioned reduction methods like thermal and chemical reduction methods, (b) repairing defects in long-chain conjugate of graphene sheets [4, 17, 38], and (c) reduction by electron transfer [28, 39]. The advantage of reducing graphene oxide by chemical reaction is that the carbon sheet structure is maintained [37]. The high carbon to oxygen ratio and the high conductivity of reduced graphene oxide have been proven by Gao et al. [32]. According to simulated results by Bagri et al. [40], if the graphene oxide sheets are covered only by functional groups without defects in the framework, the reduction can be achieved by choosing an appropriate recovery method. Defects in carbon sheets after reduction are likely to be related to oxidation during the process [35]. To overcome this problem, it has recently been recommended to use a poor oxidant in the modified Hummers method [41]. Although this reduction method has a low carbon to oxygen ratio, it produces fewer defects in graphene oxide and reduced graphene oxide has high electrical conductivity [42,43,44].
So far, numerous applications of graphene nanosheets have been reported in various fields of science [45,46,47,48,49,50,51,52,53,54]. These nanosheets containing functional groups (such as OH, COOH and C=O) are widely used for electrochemical and bioelectrochemical sensing, corrosion protection, catalytic application and energy storage. The findings demonstrate the advantage of graphene nanosheets. Magnetic graphene nanoparticles are also have many applications such as drug delivery, treatment of cancers, magnetic imaging, magnetic separation and analytical applications, making rubber and polymer composites, sensors and heterogeneous catalyst [55]. However, for introducing the magnetic property to the graphene, need to attach or composite the graphene with magnetic nanoparticles (Fe3O4) [46].
Several methods have been reported to reduce the graphene oxide with various chemical reagents that most of these reagents are toxic and flammable and some of them are not affordable. This paper focuses on the use of non hazardous, low-cost and environmentally friendly reagents to reduce graphene oxide. Achieving green and low levels of magnetic graphene is the main goal of this study. In this work, RGO firstly produced from GO in a green and safe method by using Fe2+ ions as reduction agent and then by adjusting the ratio of Fe2+ to produced Fe3+ ions during reduction of GO, the magnetic nanoparticles (Fe3O4) were precipitated on the RGO surface to synthesis the RGO–MNP. The electrochemical behavior of the prepared nanocomposites were studied and ultimately their use in a sensor for determination of lead ion were evaluated.
2 Experimental section
2.1 Materials and reagents
Graphite and other chemicals were of analytical grade, obtained from Merck or Sigma and used as supplied without further purification. The Pb(II) stock solution was prepared by dissolution of 0.0082 g of Pb(NO3)2 in 25.0 mL of double distilled water to obtain a solution of 1.0 mM Pb(II). Then, with successive dilutions, standard solutions were obtained at lower concentrations. The pH was adjusted with 10.0 mM NaOH or HCl solutions. The vessels were soaked in 3.0 M HNO3 and carefully cleaned before use to avoid contamination. To prepare the redox probe solution containing 1.0 mM [Fe(CN)6]3−/4− and 0.1 M KCl, 0.0422 g K4[Fe(CN)6], 0.3680 g K3[Fe(CN)6] and 0.7456 g KCl were weighted and adjusted in 100.0 mL. All solutions were prepared daily and kept away from light.
2.2 Synthesis of nanosheets
2.2.1 Graphene oxide (GO) nanosheets
To prepare the GO by the Hummers method [15], 5.0 g of graphite powder were added to the solution containing 7.5 mL of H2SO4, 2.5 g of K2S2O8 and 2.5 g of P2O5 at 80 °C. The dark blue mixture containing the initial GO was cooled at room temperature for 5 h. Then, to reach a neutral pH, the mixture was washed with distilled water and dried. In the next step, 2.5 g of the initial GO powder was mixed with 57.5 mL of H3PO4 at 0 °C and then 7.5 g of KMnO4 was added and stirred for 2 h. The reaction was completed by adding 6.2 mL of H2O2 and 350.0 mL of distilled water to the solution. Finally, the mixture was smooth and then washed with 10% V/V HCl to remove the concomitant ions. The remaining solid was GO nanosheets that were used to continue work.
2.2.2 Reduced GO (RGO) nanosheets
In order to reduction of GO, a green, an easy and environmentally friendly method was used for the first time. 15.0 mL of distilled water was added to 0.1500 g GO and subjected to ultrasonic wave for 30 min. In the following, 4.9702 g of FeCl2·4H2O was dissolved in 25.0 mL HCl 37% and then the GO solution was added and allowed that was stirred for 2 h under nitrogen gas. During the process, the color of the solution was black, which is due to the formation of RGO nanosheets.
2.2.3 GO–magnetic nanoparticle (GO–MNP) nanosheets
To magnetize the GO nanosheets, 0.2500 g synthesized GO was placed in 50.0 mL distilled water. The prepared suspension was added to 25.0 mL of 1.5 M NaOH at 80 °C under nitrogen gas, then 25.0 mL solution containing 0.1 M HCl, 0.5825 g FeCl3·6H2O and 0.2142 g FeCl2·4H2O was added dropwise to it. This should take at least 1 h to magnetize the GO nanosheets. In the end, synthesized GO–MNP nanosheets were separated with a magnet and washed twice with distilled water to completely neutralize. The resulted GO–MNP nanocomposite had 1:1 (W/W) ratio of GO and MNP. The advantage of this method for magnetization of GO is that can easily be separated from the solution [56].
2.2.4 RGO–magnetic nanoparticle (RGO–MNP) nanosheets
The RGO–MNP prepared in a one-step process for the first time. 4.9702 g of FeCl2·4H2O was dissolved in 0.1 M HCl solution containing 0.3 g GO and kept under argon gas and stirred for 72 h to convert Go to the RGO. After that, all the suspension was added dropwise to the 250.0 mL of 1.5 M NaOH, at 80 °C during 1 h and under nitrogen gas bubbling. After the reaction was completed, synthesized RGO–MNP nanosheets were separated by a magnet, washed twice with distilled water and dried for further experiments. The produced RGO–MNP had the ratio as 1:1 (W/W) of RGO and MNPs. In the same process RGO–MNP with ratio as 1:20 was also synthesized.
2.3 Electrode preparation
The magnetic carbon paste electrode (CPE) was used as a working electrode. For this purpose, the carbon paste was prepared from a uniform mixing of 0.7000 g of graphite powder with 0.3000 g of paraffin oil and placed inside the syringe containing a permanent magnet with a diameter and width as 5.0 and 3.0 mm, respectively. A copper wire was used as an electrical interface. After polishing the prepared CPE surface on the paper, 5.0 µL of suspension containing 10.0 mg/mL of each modifiers was placed on it and after drying, the resulting electrode was used for subsequent experiments. Scheme 1 presents the preparation steps of modified electrode.

Schematic representation of the CPE/RGO–MNP preparation and its interaction with Pb2+
2.4 Apparatus
A Philips PW 1800 instrument was used for X-ray diffraction (XRD) measurements and the spectra were performed at a voltage of 60 kV and current of 40 mA. The Fourier transform infrared (FTIR) spectra were obtained in the wave-number range of 400–4000 cm−1 using PerkinElmer Spectrum 65. The ultraviolet–visible (UV–Vis) spectra were recorded with a Cintra 404 spectrophotometer. The solutions were placed in quartz cuvettes and scanned over the range of 200–600 nm. Raman spectra of the samples were obtained by Takram P50C0R10 Raman spectrometer. A Philips EM 2085 instrument equipped with SADE was used for the transmission electron microscopy (TEM) imaging, which was carried out in air using a voltage of 100 kV and magnification of 1.8 × 105 times. Vibrating sample magnetometer (VSM) spectra were recorded by alternative gradient force magnetometer equipped with input power 1500 W and magnetic field 0.8 T (Magnetic Daghigh Kashan).
Electrochemical measurements were performed with a Metrohm 797 VA using a three-electrode assembly including a 50.0 mL glass cell, an Ag/AgCl (KCl 3.0 M) as the reference electrode, a Pt plate with a purity of 99.99% as the counter electrode and the modified CPE as the working electrode. All potentials were measured and reported vs reference electrode.
3 Results and discussion
3.1 Characterization of synthesized nanomaterials
3.1.1 XRD
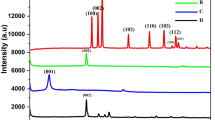
To specify the composition of the prepared nanomaterials, the intensity of the experimental peak obtained by XRD was compared with the reference peak intensity. For this purpose, the graphite, synthesized GO, RGO, GO–MNP and RGO–MNP XRD patterns were recorded (Fig. 1). The peaks appearing at 2θ of 26.5° and 12.0° in this figure related to the structure of the initial graphite and GO, respectively, which can be used to ensure the synthesis of GO due to the shift of the peak angle [43]. The expansion in the GO peak is duo to the nanostructure of the synthesized nanosheet. Regarding Fig. 1, the peak of the synthesized RGO appears at 2θ of 22.4° has less intense and wider than graphite and GO, which can be related to the small size of the synthesized nanoparticles. By studying the references [39, 43, 57], we can conclude that the synthesis of RGO is successful. Peaks appearing at 2θ of 30°, 35°, 43°, 53° and 63° in Fig. 1 related to GO–MNP and at 2θ of 31°, 36°, 44°, 58° and 62° related to RGO–MNP [57]. By comparing the XRD pattern with RGO–MNP patterns in references [58], it was found that these magnetic nanomaterials were successfully prepared.

The XRD pattern of (a) graphite, (b) GO, (c) RGO, (d) GO–MNP and (e) RGO–MNP nanosheets
3.1.2 FTIR
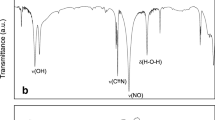
The structure of synthesized GO, RGO, GO–MNP and RGO–MNP nanomaterials were investigated by FT-IR spectroscopy and their spectra were compared with the graphite spectrum (Fig. 2). The FTIR spectrum of GO was shown strong absorption bands at 1630 cm−1 and 1740 cm−1 of carbonyl group, 3439 cm−1 of hydroxyl, 1612 cm−1 of alkene group and 1224 cm−1 of epoxy (Fig. 2, curve b). The presence of absorption bands related to functional groups containing oxygen in the GO spectrum was confirmed the correct synthesis of these nanoscale sheets [58]. The reduction of oxygen groups on the GO by Fe(II) and the formation of RGO was also confirmed by FTIR spectroscopy (Fig. 2, curve c). The strong absorption bands of functional groups such as epoxy and ketone is reduced but not eliminated, which indicates the low efficiency of these groups reduction while the absorption band of carbonyl group has been completely removed. The GO–MNP FTIR spectrum was shown in Fig. 2 curve d. The peak of the carbonyl group was shifted from 1630 to 1596 cm−1 during the magnetization process of GO. The peak appearing at 572 cm−1 refers to the stretching vibration of Fe‒O [59, 60]. As was shown in Fig. 2 curve e, the peak intensity of alkene and epoxy groups were reduced during the RGO magnetization, which may be related to the presence of Fe(II) in the magnetization reaction.

The FTIR spectra of (a) graphite, (b) GO, (c) RGO, (d) GO–MNP and (e) RGO–MNP nanosheets
3.1.3 UV–Vis
Formation of the RGO was identified by the UV–Vis spectroscopy and was compared with GO UV–Vis spectrum (Fig. 3). At the wavelength of about 230 nm, the absorption associated with π–π* transition of the aromatic bonds and another weak absorption at about 300 nm is related to the n–π* transition of the C=O band which was confirmed the synthesis of GO [37, 39]. After the reduction of GO and formation of RGO nanosheets, aromatic bonds have a redshift to 260 nm wavelength which was due to the electrical continuity of the nanosheets [44]. The amount of redshift at different reduction time was investigated and the maximum shift was occurred at 120 h (Supporting Information, Fig. S-1). The UV–Vis spectroscopy can also provide information on the solubility stability, so that if a homogeneous solution was prepared, the absorption curve will be linear as a function of concentration according to the Beer-Lambert law [61]. The UV–Vis spectra were recorded at various concentration of RGO at 260 nm (Supporting Information, Fig. S-2). The results were shown with increasing RGO concentration; absorption was also increased (R2 = 0.9980).

The UV–Vis spectra of (a) GO and (b) RGO
3.1.4 Raman
The Raman spectrum of the GO and RGO nanosheets were shown in Fig. 4. As can be seen in this figure, the intensity of the RGO band was higher than the GO band. The reason is that after the reduction reaction, due to the removal of oxygenating groups, there are defects in the nanosheets that was produced more carbon with sp3 hybridization and increased the intensity of the Raman band [37]. There are two peaks in less than 2000 cm−1 in both spectra. The first peak (D band) that was appeared at 1350 cm−1 related to defective structures edge of nanosheets or amorphous carbons with sp3 hybridization [43]. The D band extension can be related to the conversion of the carbon hybridization (sp2 to sp3) during the intense oxidation and used to track the synthesis steps of GO. The second peak (G band) at 1600 cm−1 was related to the first-degree diffusion of the E2g mode of the carbon with sp2 hybridization [43]. The ratio of D to G band intensity (ID/IG) was provided a good choice to evaluation of regular or irregular carbon nanosheets structures. This value for GO and RGO was 1.06 and 1.10, respectively. The 4% increasing for RGO in compared with GO was due to the unprocessed defects that have been created after some of the functional groups reduction.

The Raman spectra of GO and RGO nanosheets
The 2D and D + G bands at about 2700 and 2940 cm−1, respectively, in both of the GO and RGO Raman spectrum corresponded to second-order spectra. The 2D second-order Raman spectra are sensitive to the number of accumulated nanosheets on each other. It was expected that by increasing the reaction time, this ratio would be reduced due to the restoration of the framework of sp2 hybridization [39]. Observed results proofed successful synthesis of RGO from GO by using Fe(II) as a reducing agent.
3.1.5 TEM
The RGO, GO–MNP and RGO–MNP nanosheets were characterized by TEM. As can be seen in Fig. 5, the presence of graphene nanosheets in the RGO have relatively good transparency and were placed on each other. After magnetizing the GO and RGO nanosheets and producing the GO–MNP and RGO–MNP, respectively, Fe3O4 nanoparticles were randomly distributed on these nanosheets (black area in Fig. 5). The Fe3O4 nanoparticles in the RGO–MNP have accumulated alongside each other and located as well as between the nanosheets and have created a combination like a hamburger.

The TEM images of a RGO, b GO–MNP and c RGO–MNP. The inset in b is SAED pattern obtained at the GO–MNP
3.1.6 VSM
VSM has used to evaluate the magnetic properties of the GO–MNP and RGO–MNP (1:1 and 1:20) as a function of the external magnetic field applied (Fig. 6) and the results are presented in Table 1. The results have shown the GO–MNP and especially RGO–MNP with a ratio of 1:20 have a good response to the magnetic field and have superparamagnetic properties [62]. The superparamagnetic property has caused these synthesized nanosheets in this work to have not magnetic properties after removing the magnetic field and will interfere with each other. This property is very important and useful.

The VSM spectra of (a) RGO–MNP with 1:20, (b) RGO–MNP with 1:1 and (c) GO–MNP with 1:1 ratio of Fe3O4: graphene
3.2 Electrochemical investigation
Figure 7 shows the cyclic voltammograms (CVs) of the CPE before (a) and after modification by the GO (b), RGO (c), GO–MNP (d), and RGO–MNP (e) in the presence of the [Fe(CN)6]3−/4− probe. By modifying the CPE surface by GO, the insulation layer forms on the surface of the electrode, which was reduced the Ip and increased the ΔEp. After modifying the surface of the CPE by GO–MNP, the Ip has increased by 2 and 25 times relative to the CPE and CPE/GO, respectively. This increase in the Ip can be attributed to the presence of Fe3O4 magnetic nanoparticles at the surface of graphene nanosheets and providing more conductivity in these [63]. As shown in Fig. 7, the modified CPE by RGO has a much higher capacitance current than CPE and CPE/GO (the Ip has been increased by about 5 and 150 times, respectively), which indicates that the superconducting property of the RGO nanosheets. Due to the presence of Fe3O4 magnetic nanoparticles in the CPE/RGO–MNP and superconductivity properties, the Ip was reduced in CPE/RGO–MNP in compared to the CPE/RGO (Fig. 7, curves d and e).

The CVs obtained on the (a) CPE, (b) CPE/GO, (c) CPE/RGO, (d) CPE/GO–MNP and (e) CPE/RGO–MNP in 1.0 mM [Fe(CN)6]3−/4− containing 0.1 M KCl
To study the electrochemical behavior of the reduction–oxidation process of [Fe(CN)6]3−/4− at the electrode surface, CVs of the CPE, CPE/GO, CPE/RGO, CPE/GO–MNP and CPE/RGO–MNP at different scanning rates were recorded in 1.0 mM of [Fe(CN)6]3−/4− and 0.1 M KCl. As can be seen in Figs. 8 and S-3, anodic and cationic currents in all electrodes were increased linearly with the square root of scan rate (Table 2) that suggested the reaction of the redox probe at all electrodes was controlled by the diffusion process. According to Fig. 8, for all the electrodes used in this work, with an increase in the potential scanning rate from 50 to 300 mV, a displacement of about 60 mV in potential appears. The observed behavior indicates the quasi-reversible reaction of the redox at the surface of all electrodes.

The CVs obtained for the a CPE/GO, b CPE/RGO, c CPE/GO–MNP and d CPE/RGO–MNP in 1.0 mM [Fe(CN)6]3−/4− containing 0.1 M KCl at different scan rates. Insets show the cathodic and anodic peak current versus ν1/2
The presence of mound and fovea at the electrode surface increases its real surface relative to the geometric surface and thus increases the roughness factor (real area/geometric area), which increases the catalytic response of the electrode to reduction reactions [64]. The real electrode surface area was calculated by Randle–Sevcik equation [65], Ip = 2.69 × 105n3/2CD1/2Aν1/2, in which n is the number of the transferred electron, C is the concentration of electroactive species, D is the diffusion coefficient, A is the real electrode surface area and ν is the potential scanning rate. The real surface area of the CPE, CPE/GO, CPE/RGO, CPE/GO–MNP and CPE/RGO–MNP was calculated using the Ip versus ν1/2 curves (Fig. 8) and Randle-Sevick equation and presented in Table 2. The presence of the GO groups on the surface of the CPE and its coating effect causes the real surface area of the CPE/GO to decrease 10% compared with the CPE. The real surface area of the CPE/RGO was about 2 and 20 times higher than the CPE and CPE/GO, respectively, which can be attributed to the RGO nanosheets in compared with the GO during the modification process. According to Table 2 and Fig. 8, it can be concluded that the increase of 25 and 20 times at the CPE/GO–MNP current and real surface area, respectively, and a decrease of 1.5 and increase four times at the CPE/RGO–MNP current and real surface area were due to the presence of iron magnetic nanoparticles on nanosheets.
3.3 Analytical characteristics
The interaction of two valance cations with CPE/GO–MNP and CPE/RGO–MNP was investigated. Square wave voltammograms (SWVs) of mentioned electrodes after preconcentration for 25 min in 1.0 M each of Pb2+, Co2+, Ni2+, Zn2+, Cd2+ and Cu2+ solution were recorded in 0.5 M HCl (Fig. S-4). The analysis of these graphs was shown the CPE/RGO–MNP has a better response to Cu2+ and Pb2+, and the CPE/GO–MNP has a better response to Pb2+ than other ions. Therefore, for comparison of these electrode performances, Pb2+ ion was selected. Due to cumulative toxicant of Pb and its effect on multiple body systems, its determination and elimination from water is important [66, 67].
3.3.1 Response characteristics of the CPE and modified CPE to Pb2+
The SWV of CPE, CPE/MNP, CPE/GO, CPE/RGO, CPE/GO–MNP and CPE/RGO–MNP after preconcentration in a 1.0 mM solution of Pb2+ was recorded in 0.5 M HCl solution (Fig. 9). As is clear from the figure, the unmodified electrode has no interaction with Pb2+ and was not able to preconcentrate this ion. The modification of the electrode surface with Fe3O4 nanoparticles was led to preconcentrate Pb2+ and a slight stripping current at the CPE/MNP surface. It is expected that the modification of the CPE surface by GO with abundant functional groups (epoxy, hydroxyl and carboxylic acid) of the surface was led to preconcentrate Pb2+ and made high stripping current. As can be seen in Fig. 9 curve c, a relatively high current of about 0.9 mA is due to the accumulation of Pb2+ at the CPE/GO. It is expected that, by reduction the GO to RGO, the functional groups containing oxygen of the GO eliminated and therefore RGO ability to collect Pb2+ decreased. The Ip of CPE/RGO was higher than the CPE/GO which can be attributed to better electronic properties (higher conductivity) of the RGO in compared to the GO. The presence of Fe3O4 magnetic nanoparticles at the CPE/GO–MNP and CPE/RGO–MNP due to electrocatalytic properties and stripping of Pb2+ has led to increase in the Ip at both surfaces (Fig. 9, curves e and f).

The SWV obtained in 0.5 M HCl solution on the (a) CPE, (b) CPE/MNP, (c) CPE/GO, (d) CPE/RGO, (e) CPE/GO–MNP and (f) CPE/RGO–MNP after preconcentration in 1.0 mM Pb2+ solution
3.3.2 Optimization of experimental conditions
The effective parameters on the interaction between Pb2+ and modified electrodes (CPE/GO–MNP and CPE/RGO–MNP) were optimized. The pH of 1.0 mM Pb2+ solution was changed in the range of 1.5–5.0 and after preconcentration for 15 min, the SWV of the electrodes was recorded in 0.5 M HCl (Fig. 10). The Ip was maximum at pHs 3.0 and 3.5 for CPE/GO–MNP and CPE/RGO–MNP, respectively. According to the solubility product constant of Pb(OH)2 (2.5 × 10−16), Pb2+ being to precipitate at pH 7.7 and removed from the solution. Competition between active groups at the electrodes surface with OH‒ for collecting Pb2+ and saturation surface of the electrodes with Pb2+ were caused the Ip was decreased at pHs lower than 3.0 and 3.5 for CPE/GO–MNP and CPE/RGO–MNP, respectively.

The SWVs obtained for the a CPE/GO–MNP and b CPE/RGO–MNP in 0.5 M HCl after preconcentration in 1.0 mM Pb2+ for 15 min at different pH. Insets show the variation of Ip versus pH of preconcentration solution
Another the investigated factor was the effect of preconcentration time on the CPE/GO–MNP and CPE/RGO–MNP response. Preconcentration was performed in 1.0 mM Pb2+ solution for both electrodes at different times. The SWVs of the electrodes in the 0.5 M HCl at pH 3.0 for CPE/GO–MNP (Fig. 11a) and 3.5 for CPE/RGO–MNP (Fig. 11b) were recorded. As shown in this figure, with increasing of preconcentration time, the intensity of Ip was increased and reached to a maximum value at 10 min for CPE/GO–MNP and 25 min for CPE/RGO–MNP.

The SWVs obtained for the a CPE/GO–MNP and b CPE/RGO–MNP in 0.5 M HCl after preconcentration in 1.0 mM Pb2+ at pH 3.0 and 3.5, respectively, at different time. Insets show the variation of Ip versus preconcentration time
The next factor that affects the electrode response is the amount of GO–MNP and RGO–MNP were used to modify the CPE. To evaluate this factor, the CPE was modified with 1, 3, 5, 7, 10, 13 and 15 µL of 10 mg/mL GO–MNP suspension, and with 1, 3, 5, 7 and 10 µL of 10 mg/mL RGO–MNP suspension. The SWVs of the CPE/GO–MNP and CPE/RGO–MNP after preconcentration of Pb2+ were recorded in 0.5 M HCl (Fig. 12). As can be seen from this figure, the maximum Ip observed for CPE modified by 10 µL of GO–MNP and 7 µL of RGO–MNP.

The SWVs obtained in 0.5 M HCl on the a CPE/GO–MNP and b CPE/RGO–MNP with different modifier amount of GO–MNP or RGO–MNP suspension. Preconcentration condition: 1.0 mM Pb2+ solution at pH 3.0 for 10 min and pH 3.5 for 25 min for the CPE/GO–MNP and CPE/RGO–MNP, respectively. Insets show the variation of Ip versus amount of modifier
3.3.3 Optimization of instrumental conditions
The effect of the instrumental factors including square wave frequency, amplitude potential and step potential on the CPE/GO–MNP and CPE/RGO–MNP response were investigated. In each case, two factors were kept constant and one factor optimized (Supporting Information S-5 to S-7). The SWVs of CPE/GO–MNP after preconcentration in a 1.0 mM Pb2+ solution at pH 3.0 for 10 min and CPE/RGO–MNP after preconcentration in 1.0 mM Pb2+ solution at pH 3.5 for 25 min were recorded (Figs. S-5 to S-7). Regarding the recorded voltammograms, square wave frequency as 40 Hz, amplitude potential as 35 mV, and step potential as 10 mV for CPE/GO–MNP, and square wave frequency as 100 Hz, amplitude potential as 25 mV, and step potential as 5 mV for CPE/RGO–MNP were chose as the best instrumental conditions.
3.3.4 Calibration curve
The CPE/GO–MNP and CPE/RGO–MNP were placed in Pb2+ solutions with the concentration of 1 × 10−9, 5 × 10−9, 1 × 10−8, 5 × 10−8, 1 × 10−7, 5 × 10−7, 1 × 10−6, 5 × 10−6, 1 × 10−5, 5 × 10−5, 1 × 10−4, 5 × 10−4 and 1 × 10−3 M in the optimized experimental conditions. Then, SWVs were recorded in 0.5 M HCl solution and optimized instrumental conditions (Fig. 13). By increasing the concentration of Pb2+ ions in the preconcentration solution, the Ip was increased (Fig. 13, insets) and the relationship between Ip and concentration was investigated (Table 3). Regarding the observed trend, it can be concluded at low concentration of Pb2+, the functional groups that have a large formation constant (Kf) with Pb2+ have adsorbed this ion. By increasing the concentration of Pb2+, functional groups with small Kf also being to adsorb Pb2+ ion from the solution and it was caused the slope of the curve had an incremental trend.

The SWVs obtained in 0.5 M HCl on the a CPE/GO–MNP and b CPE/RGO–MNP after immersion in standard solutions of Pb2+ at pH 3.0 and 3.5, respectively. Calibration curves are presented as insets
The interference of some cations on SWV response of 1 mM of Pb2+ was investigated on both electrode. Co2+, Zn2+, Ni2+, Mg2+, and Cd2+ in 100-fold, K+, and Na+ in 1000-fold did not effect on the signal of Pb2+. Also, the interference of Cu2+ towards Pb2+ was performed. Due to well-separate Pb2+ and Cu2+ voltammetric waves (> 300 mV) at the both electrodes, the observed faradaic current for Cu2+, didn’t interfere with Pb2+.
The figures of merit were calculated for the CPE/GO–MNP and CPE/RGO–MNP. The detection limit (DL) of the CPE/GO–MNP and CPE/RGO–MNP for determination of Pb2+ using the obtained voltammograms was calculated as 8.13 × 10−10 and 3.07 × 10−9 M, respectively. The relative standard deviation (RSD) of the CPE/GO–MNP and CPE/RGO–MNP for four times the measuring of Pb2+ at a constant concentration of 1.0 mM was 5.4% and 4.2%, respectively, indicating high repeatability of the electrodes for measuring of Pb2+. In order to investigate the reproducibility of the electrodes, the RSD for four different electrodes in measuring the Pb2+ at a specific concentration was obtained as 6.4% and 6.1% for CPE/GO–MNP and CPE/RGO–MNP, respectively. Observed results showed the high reproducibility of prepared modified CPE electrodes. The stability of the electrodes also examined. A series of 30 repetitive voltammetric measurements has been carried out for 1.0 mM Pb2+. The coefficient of variation was found to be 5.5% and 4.4% for the CPE/GO–MNP and CPE/RGO–MNP, respectively. This results indicating that the electrodes are stable and does not undergo by fouling products.
4 Conclusion
In this research, graphene oxide was prepared by the Hummers method, then using green and easy method deduced to graphene for the first time. It was reduced by Fe2+ with an electron transfer mechanism, become to graphene nanosheets and characterized by spectroscopic and electrochemical methods. Within this effective method, can synthesize GO–MNP and RGO–MNP in one step. Also, the behavior of CPE/RGO–MNP and CPE/GO–MNP was investigated in different pHs, which 3.5 and 3.0 were selected as the optimum pH, respectively. In order to analytical characterization, CPEs modified with magnetic composites were prepared and used for the determination of Pb2+ ion. A linear calibration curve for CPE/RGO–MNP and CPE/GO–MNP from 1.0 × 10−9‒1.0 × 10−3 M Pb2+ (R2 = 0.9960) and 1.0 × 10−9‒5.0 × 10−3 (R2 = 0.9946), with a detection limit as 3.07 × 10−9 and 8.13 × 10−10 were obtained, respectively. The results were shown CPE/RGO–MNP had more interaction with Pb2+ ion and better performance in the determination of this ion than CPE/GO–MNP.
References
M. Bagherzadeh, A. Farahbakhsh, Graphene Materials: Fundamental and Emerging Applications (Wiley, New York, 2015), p. 25
C. Lee, X. Wei, J.W. Kysar, J. Hone, Science 321, 385 (2008)
I.-Y. Jeon, Y.-R. Shin, G.-J. Sohn et al., Proc. Natl. Acad. Sci. USA 109, 5588 (2012)
Z.-Y. Juang, C.-Y. Wu, A.-Y. Lu et al., Carbon 48, 3169 (2010)
C. Schafhaeutl, Philos. Mag. 16, 570 (1840)
B.C. Brodie, Philos. Trans. R. Soc. Lond. 149, 249 (1859)
T. Szabó, O. Berkesi, P. Forgó et al., Chem. Mater. 18, 2740 (2006)
J.I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso, J. Tascon, Langmuir 25, 5957 (2009)
C. Gómez-Navarro, J.C. Meyer, R.S. Sundaram et al., Nano Lett. 10, 1144 (2010)
D. Pandey, R. Reifenberger, R. Piner, Surf. Sci. 602, 1607 (2008)
D.A. Dikin, S. Stankovich, E.J. Zimney et al., Nature 448, 457 (2007)
Y. Liu, B. Xie, Z. Zhang, Q. Zheng, Z. Xu, J. Mech. Phys. Solids 60, 591 (2012)
B. Marinho, M. Ghislandi, E. Tkalya, C.E. Koning, G. de With, Powder Technol. 221, 351 (2012)
D.R. Dreyer, S. Park, C.W. Bielawski, R.S. Ruoff, Chem. Soc. Rev. 39, 228 (2010)
W.S. Hummers Jr., R.E. Offeman, J. Am. Chem. Soc. 80, 1339 (1958)
S. Stankovich, D.A. Dikin, R.D. Piner et al., Carbon 45, 1558 (2007)
C. Mattevi, G. Eda, S. Agnoli et al., Adv. Func. Mater. 19, 2577 (2009)
M.J. McAllister, J.-L. Li, D.H. Adamson et al., Chem. Mater. 19, 4396 (2007)
G. Xin, W. Hwang, N. Kim, S.M. Cho, H. Chae, Nanotechnology 21, 405201 (2010)
Y. Zhu, S. Murali, M.D. Stoller, A. Velamakanni, R.D. Piner, R.S. Ruoff, Carbon 48, 2118 (2010)
I.K. Moon, J. Lee, R.S. Ruoff, H. Lee, Nat. Commun. 1, 73 (2010)
L. Jiao, X. Wang, G. Diankov, H. Wang, H. Dai, Nat. Nanotechnol. 5, 321 (2010)
X. Fan, W. Peng, Y. Li et al., Adv. Mater. 20, 4490 (2008)
X. Zhou, J. Zhang, H. Wu, H. Yang, J. Zhang, S. Guo, The Journal of Physical Chemistry C 115, 11957 (2011)
P.V. Kamat, Chem. Rev. 93, 267 (1993)
Y.H. Ng, A. Iwase, A. Kudo, R. Amal, J. Phys. Chem. Lett. 1, 2607 (2010)
S.J. An, Y. Zhu, S.H. Lee et al., J. Phys. Chem. Lett. 1, 1259 (2010)
M. Zhou, Y. Wang, Y. Zhai et al., Chemistry 15, 6116 (2009)
S. Dubin, S. Gilje, K. Wang et al., ACS Nano 4, 3845 (2010)
G. Demazeau, J. Mater. Chem. 9, 15 (1999)
H. Wang, J.T. Robinson, X. Li, H. Dai, J. Am. Chem. Soc. 131, 9910 (2009)
W. Gao, L.B. Alemany, L. Ci, P.M. Ajayan, Nat. Chem. 1, 403 (2009)
X. Wang, L. Zhi, K. Müllen, Nano Lett. 8, 323 (2008)
H. Chen, M.B. Müller, K.J. Gilmore, G.G. Wallace, D. Li, Adv. Mater. 20, 3557 (2008)
G. Eda, G. Fanchini, M. Chhowalla, Nat. Nanotechnol. 3, 270 (2008)
D. Yang, A. Velamakanni, G. Bozoklu et al., Carbon 47, 145 (2009)
S. Pei, J. Zhao, J. Du, W. Ren, H.-M. Cheng, Carbon 48, 4466 (2010)
V. López, R.S. Sundaram, C. Gómez-Navarro et al., Adv. Mater. 21, 4683 (2009)
Z.-J. Fan, W. Kai, J. Yan et al., ACS Nano 5, 191 (2010)
A. Bagri, C. Mattevi, M. Acik, Y.J. Chabal, M. Chhowalla, V.B. Shenoy, Nat. Chem. 2, 581 (2010)
Y. Xu, K. Sheng, C. Li, G. Shi, J. Mater. Chem. 21, 7376 (2011)
X. Zhao, J. Wang, F. Wu et al., J. Hazard. Mater. 173, 102 (2010)
C. Zhu, S. Guo, Y. Fang, S. Dong, J.B. Liu, S.H. Fu, B. Yuan, Y.L. Li, Z.X. Deng, J. Am. Chem. Soc. 132, 4490 (2010)
Y. Wang, Z. Shi, J. Yin, ACS Appl. Mater. Interfaces. 3, 1127 (2011)
M. Bagherzadeh, M. Heydari, Analyst 138, 6044 (2013)
M. Bagherzadeh, M. Amrollahi, S. Makizadeh, RSC Adv. 5, 105499 (2015)
N. Ahmadi, A. Nemati, M. Bagherzadeh, J. Alloys Compd. 742, 986–995 (2018)
M. Bagherzadeh, S. Mozaffari, M. Momeni, Anal. Methods 7, 9317 (2015)
Z. ShamsGhahfarokhi, M. Bagherzadeh, E. GhiamatiYazdi, A. Teimouri, Anti-Corros. Methods Mater. 65, 249–262 (2018)
M. Bagherzadeh, Z.S. Ghahfarokhi, E.G. Yazdi, RSC Adv. 6, 22007 (2016)
E.G. Yazdi, Z.S. Ghahfarokhi, M. Bagherzadeh, New J. Chem. 41, 12470 (2017)
A.N. Golikand, M. Bagherzadeh, Z. Shirazi, Electrochim. Acta 247, 116 (2017)
M. Nasrollahzadeh, S.M. Sajadi, A. Rostami-Vartooni, M. Alizadeh, M. Bagherzadeh, J. Colloid Interface Sci. 466, 360 (2016)
M. Nasrollahzadeh, M. Maham, A. Rostami-Vartooni, M. Bagherzadeh, S.M. Sajadi, RSC Adv. 5, 64769 (2015)
Y.-F. Yang, F.-Y. Meng, X.-H. Li et al., J. Nanosci. Nanotechnol. 19, 7517 (2019)
M. Bagherzadeh, S. Ansari, F. Riahi, A. Farahbakhsh, Int. J. Electrochem. 2013, 1–10 (2013)
H. Zhang, M.E. Meyerhoff, Anal. Chem. 78, 609 (2006)
C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska, L. Niu, Langmuir 25, 12030 (2009)
O. Cozar, N. Leopold, C. Jelic et al., J. Mol. Struct. 788, 1 (2006)
M.C. D’Antonio, A. Wladimirsky, D. Palacios et al., J. Braz. Chem. Soc. 20, 445 (2009)
S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S.T. Nguyen, R.S. Ruoff, ACS Nano 2, 572 (2008)
J.-J. Yuan, S. Armes, Y. Takabayashi et al., Langmuir 22, 10989 (2006)
J. Yan, T. Wei, B. Shao et al., Carbon 48, 1731 (2010)
B. Kumara Swamy, J. Maye, C. Vannoy, M. Schell, J. Phys. Chem. B 108, 16488 (2004)
A.J. Bard, L.R. Faulkner, J. Leddy, C.G. Zoski, Electrochemical Methods: Fundamentals and Applications, vol. 2 (wiley, New York, 1980)
P. Gu, S. Zhang, C. Zhang et al., Dalton Trans. 48, 2100 (2019)
W. Chen, Z. Lu, B. Xiao et al., J. Clean. Prod. 211, 1250 (2019)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10854_2019_2407_MOESM1_ESM.docx
SWVs regarding the interaction of divalent cations with CPE/GO–MNP and CPE/RGO–MNP, Optimization of instrumental conditions, UV–Vis spectra at different deposition time and concentration of RGO, CVs regarding the effect of scan rate on CPE response. Supplementary material 1 (DOCX 1637 kb)
Rights and permissions
About this article

Cite this article
Bagherzadeh, M., Jabouri-Abassi, M. & Akrami, Z. One-step synthesis of reduced graphene oxide and magnetic graphene: characterization and its application in electrochemical detection of lead (II) ions. J Mater Sci: Mater Electron 30, 20229–20242 (2019). https://doi.org/10.1007/s10854-019-02407-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-019-02407-5