
Abstract
Enhanced green and red upconversion emissions under 980 nm excitation are observed in Ba0.85−xCa0.15TiO3:0.75%Er3+/xIn3+ lead-free ferroelectric ceramics which were synthesized by solid state reaction method. With increasing In3+ ions doping level from x = 0% mol the green and red emission intensities increase and reach the maximum at x = 1.75% mol. The maximum intensity of green emission at 550 nm is 20 times larger than In3+ ions undoped samples, which is unprecedented for Er3+-doped perovskite ferroelectric materials. Based on the ultraviolet-visible-near infrared absorption spectra, the enhancement of the luminescence intensity of the A-site In3+ doped Ba0.85−xCa0.15TiO3:0.75%Er/xIn ceramics is attributed to In3+-doping-induced relative increase in isolate Er3+ and decrease in the clustered-Er3+. The effects of In3+ doping on the crystal structure, dielectric, ferroelectric, piezoelectric properties of the ceramics were also studied and discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Upconversion luminescence is an anti-Stokes type emission, in which the wavelength of emission light is shorter than the excitation wavelength, due to the sequential absorption of two or more photons [1]. Rare-earth ions doped perovskite ferroelectric materials, owing excellent ferroelectric, piezoelectric and luminescence properties, are currently attracting extensive interest and have potential applications in the field of sensors, optoelectronic and display devices etc. [2,3,4]. Improving luminescence efficiency of rare-earth doped materials is an important precondition for practical device applications, but there are still few studies on upconversion luminescence efficiency of perovskite ferroelectrics until now.
Er3+ ions are commonly used as emission center in upconversion luminescent materials because of its special energy level which has many fine splitting and metastable energy levels. The 4f electrons of Er3+ ions enable easy excitation in a wide photoluminescence emission range from visible to mid-infrared. Er3+ doped ferroelectrics can be excited by both 980 and 1550 nm infrared and show green emission at 530, 550 nm and red emission at 670 nm in visible light range [5,6,7]. Many studies have been carried out on the upconversion luminescence of different Er3+-doped materials, especially perovskite-type ferroelectrics, such as Er3+-doped PbZr0.52Ti0.48O3, Pb(Mg1/3Nb2/3)O3–PbTiO3–Pb(Yb1/2Nb1/2)O3, (K0.48Na0.48Li0.04)(Nb0.96Bi0.04)O3, LiNbO3, BaTiO3, Ba0.95Sr0.05Zr0.1Ti0.9O3, and Na0.5Bi0.5TiO3 etc. [8,9,10,11,12,13,14]. And the enhancement effect of Er3+/Yb3+ co-doped and Tm3+/Yb3+ co-doped in transparent phosphate glass–ceramics have been studied [15, 16]. Although there are many studies on electrical properties and luminescence properties of Er3+ doped ferroelectrics, only a low luminescence efficiency could be obtained. It is necessary to further improve luminescence efficiency of Er3+-doped perovskite ferroelectrics.
Recently, Li et al., Noto et al. and Qian et al. reported that the In3+ doping can improve the emission intensities of Ga2O3:Cr3+, CaTiO3:Pr3+ and LiNbO3:Er3+/Yb3+, respectively [5, 17, 18]. Zhang et al. and Han et al. found that the emission intensities of Y2.94(Al1−x−yGaxIny)5O12:Er3+ (x + y = 0.15) and NaY0.95−xYb0.03Er0.02F4:In3+ increase by In3+ doping [19, 20]. Our previous studies also demonstrated that In3+-ions-doped perovskite-type Ba0.85Ca0.15TiO3:0.75%Er/xIn achieved 10 times increase in green band emission intensity prominently [6, 7]. To further improve the upconversion photoluminescent properties of Ba0.85Ca0.15TiO3:0.75%Er, we prepared a series of A-site In3+-ions-doped Ba0.85−xCa0.15TiO3:0.75%Er3+ (B1−xCT:0.75%Er) ceramics and studied the ferroelectric, piezoelectric, dielectric, structural and upconversion photoluminescent properties. We found that the green upconversion emission of B1−xCT:0.75%Er/xIn could be dramatically enhanced by 20 times through nominal A-site In3+ doping. Ultraviolet-visible-near infrared absorption spectra reveals that the In3+ doping increases the relative volume fraction of isolate In3+ ions, which favors the enhancement of green emission.
2 Experiment
The Ba0.85−xCa0.15TiO3:0.75%Er3+/xIn3+ (0 ≤ x ≤ 2%) ceramics were prepared by the conventional solid-state method using the high purity raw materials of BaCO3 (99.8%), CaCO3 (99.5%), TiO2 (99.9%), Er2O3 (99.9%) and In2O3 (99.99%). These weighed powders were ball mixed in the alcohol medium for 24 h, then dried at 100 °C and calcined at 1200 °C for 4 h. The weight of the raw material and the weight of the pre-sintered precursor are showed in Table 1. The powders were ball milled again for another 24 h, and pressed into pellets with 10 mm in diameter and 1 mm in thickness. Samples were sintered at 1350 °C in the air for 3 h. For electrical measurements, top and bottom surfaces of the samples were coated with sliver paste and sintered at 600 °C for 2 h.
X-ray diffraction with a Cu Kα radiation (Bruker D8 Advance) was used to characterize the phase structure of the samples, at a 40 kV tube voltage and a 40 mA tube current. Microstructures of the samples were characterized by a field emission scanning electronics microscope (FE-SEM, S-4800, HITACHI). Dielectric properties were measured between − 200 °C-150 °C using an Agilent E4980A Precision LCR analyzer at 100 kHz. Ferroelectric hysteresis loops were measured at room temperature using a Precision Multiferroic test system (Radiant Technologies, Inc. USA). The piezoelectric coefficient d33 was measured using a quasi-static d33 meter (ZJ-4AN). A spectrofluorometer (Edinburgh Instruments FS5, UK) was used to measure the upconversion emission spectra of the ceramics samples, and a laser diode operating at a fixed pump power of 49 mW and a beam waist of 1 mm was used to provide 980 nm laser source. The absorption spectra were measured by an ultraviolet-vis-near infrared (UV-Vis-NIR) absorption spectrophotometer (Cary 5000).
3 Results and discussion
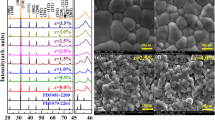
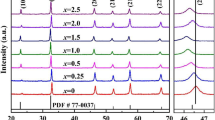
The phase structures of the samples were studied by analyzing their XRD patterns and temperature-dependent dielectric constant (εr versus T) curves. Figure 1a displays XRD patterns of B1−xCT:0.75%Er/xIn samples. All diffraction peaks can be indexed according to those of BaTiO3. PDF#79-2264 and PDF#81-2200 are standard XRD patterns of tetragonal phase (T) and orthorhombic phase (O) of BaTiO3, respectively. All samples with various In3+ content show perovskite structure and no impurity phase can be detected. The phases of two diffraction peaks were unchanged with different In3+ content in the 2θ range from 45°–46° (Fig. 1b). All samples are pure tetragonal phase at room temperature. Characteristic peaks strength was changed slightly, which represents the degradation of crystallization under high doped. With the doping of In3+ ions, the grain size of the samples did not change significantly, according to the SEM images (Fig. 1c). Figure 2a shows the curves of dielectric properties (εr and tan δ) against temperature. In the measured temperature region from − 200 to 150 °C a strong dielectric peak and three loss tan δ peaks appear. The dielectric peak corresponds to the cubic-to-tetragonal phase transition, and three loss tan δ peaks correspond to the cubic-to-tetragonal, tetragonal-to-orthorhombic, and orthorhombic-to-rhombohedral phase transitions, respectively [6, 7, 21]. With increasing x from 0 to 2%, the Curie Temperature TC decreases linearly from 138 to 118 °C, similar to previously reported results [6, 7]. The reason for the decrease of TC may probably arise from compositional and structural disorder due to the difference in the valences and ionic radii between Ba2+ and In3+. Moreover, the possible replacement of Ba2+/Ca2+ ions by Er3+ ions would lead to oxygen and titanium vacancies, which could also contribute to the decrease of TC [22,23,24,25]. According to the dielectric and loss tanδ against the temperature data, we establish the phase diagram of the B1−xCT:0.75%Er/xIn, as shown in Fig. 2b. It is noted that the tetragonal-to-orthorhombic transition temperature (TO−T) is relativly insensitive to the In3+ content for 0 to 2%. All samples exhibit tetragonal phase at room temperature, which is consistent with the XRD results.

a XRD pattern of the B1−xCT:0.75%Er/xIn (0 ≤ x ≤ 2%) ceramics. b Enlarge XRD patterns from 45° to 46°. c SEM images of the x = 0%, 1%, and 2% samples

a Polarization–electric field hysteresis loops of the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics. b, c 2Pr and d33 as a function of In3+ content for the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics. d Temperature dependence of the dielectric constant for the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics. Inset is the dielectric loss against temperature for the samples. e Phase diagram of the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics
Figure 2c shows the polarization–electric (P-E) hysteresis loops of the B1−xCT:0.75%Er/xIn (0 ≤ x ≤ 2%) ceramics whose remanent polarization (2Pr) are shown in Fig. 2d. With increasing In3+ content from x = 0%, 2Pr increases slightly and reaches a maximum of 23.5 µC/cm2 at x = 0.5%. With further increase in the In3+ content 2Pr decreases rapidly to a minimum of 11.9 µC/cm2 at x = 2.0%. As mentioned above, the replacement of A-site Ba2+ by In3+ would result in charge balance broken, formation of titanium and oxygen vacancies [22, 23]. It has been reported that oxygen vacancies induce defect dipoles [26, 27] which would suppress ferroelectric domain switching due to the pinned effect, resulting in an enhancement of the remnant polarization (2Pr) in the low x range 0%≤x≤0.5% [6, 28, 29]. However, a further increase in the In3+ content leads to a decrease in the remnant polarization. Excessive In3+ doping induced more oxygen vacancies cause the unit cell serious distortion and dissymmetry, thus ferroelectric domains breaking and leakage current take place [28, 30]. Additionally, the piezoelectric coefficient d33 continuous decreases from ~ 130 to ~ 50 pC/N (Fig. 2e), with increasing In3+ content from 0 to 2%. All previously discussed effects including increasing In3+ ions, oxygen vacancies, pinning effect, leakage current and ferroelectric domains destruction, are negative factors of piezoelectric properties [31, 32]. These reasonable explanations are consistent with the experimentally observed variation trend of 2Pr and d33.
Figure 3a shows the upconversion luminescence spectra of the B1−xCT:Er/xIn using 980 nm laser excitation at room temperature. Each spectrum shows three emission bands, two strong green emission bands at 530 and 550 nm, and one weak red emission band at 670 nm, corresponding to the 2H11/2/4S3/2 → 4I15/2 (green) and 4F9/2 → 4I15/2 (red) transitions of Er3+ ions, respectively. The location and intensity of the peaks are consistent with previous reports [6, 7, 33]. The emission intensity of all the three peaks are plotted against the In3+ content in the inset of Fig. 3a, where one can find that the maximum emission intensity of each peak appears at x = 1.75%. Notably, the maximum emission intensity at 550 nm is approximately 20 times larger than that of In3+ undoped samples. For the 530 and 670 nm emission peaks, the In3+ doping enhances the emission intensity by 16 and 20 times, respectively, as compared with those of In3+ undoped one. The emission intensities of all peaks (530, 550, 670 nm) reach the maximum at x = 1.75% and then decrease with further increases in the In3+ content. In3+-doping-induced changes in the energy level could improve the luminence intensity within a certain In3+ content, heavy In3+ doping would induce quenching effect and result in quick degeneration of emission intensity. As discussed in the above section, the In3+ doping induces oxygen vacancies which would induce defect levels, which act as traps for electrons populated in the conduction band and suppress energy transition to the excited levels of Er3+ ions [19]. In this work, 20 times enhancement in the emission intensity was unprecedented, comparing with previous reports [6, 7].

a The upconversion emission spectra of the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics. Inset is the emission intensity at 530, 550 and 670 nm as a function of the In3+ content. b The emission spectra of the B1−0.0175CT:Er/1.75%In ceramics under the excitation of different pump power. Inset is the Ln–Ln plot of the emission intensity versus pump power. c Simplified energy level diagram for the Er3+ ions upconversion mechanisms. d The absorption spectra of the B1−xCT:Er/xIn (0 ≤ x ≤ 2%) ceramics. Inset is the emission intensity at 550 nm and the absorption cross section of the 2H11/2 level as a function of the In3+ content
To further investigate the effect of In3+ doping on the luminescence mechanism of B1−xCT:Er/xIn ceramics, we plot the emission spectra under different excitation pump power in Fig. 3b and measure the emission intensity (I) as a function of pump power (P) for the sample of x = 1.75%. As is reported in previous research works, [34, 35] the number of photons (n) required to populate the upper emitting state can be described by the relationship I ∝ Pn. The estimated slopes (n) from the linear fits of ln(I) versus ln(P) are 2.03, 1.97 and 1.86, corresponding to the emission bands for 530, 550 and 670 nm, respectively. It indicates that the two-photon excitation mechanism dominates the upconversion luminescence of Er3+ ions in In3+ doped BCT:0.75Er% ceramics.
Figure 3c shows the simplified energy level diagram of the upconversion luminescence under 980 nm excitation [6, 7, 30, 33, 36]. According to the schematic diagram, 4f electrons of Er3+ ions are excited from the ground state 4I15/2 to long-lived excited state 4I11/2 through the ground state absorption (GSA) by the 980 nm excitation. Under sustaining excitation, the electrons at 4I11/2 level absorb the second photon energy and transition to higher energy levels of 4F7/2 through excited state absorption (ESA). Consequently, the electrons at 4F7/2 level relax to 2H11/2, 4S3/2 and 4F9/2 levels without radiation, and then radiative transition to the ground state, 2H11/2/4S3/2 → 4I15/2 (green) and 4F9/2 → 4I15/2 (red), occurs. In addition, the multiphonon relaxation (MPR) during which electrons from excited levels jump into low level without radiation results in the decrease of photons absorption efficiency. Furthermore, the cross relaxation (CR) between two adjacent Er3+ ions is associated with the In3+ content and will be discussed in following sections.
The UV-VIS-NIR absorption spectra for selected samples in the wavelength ranging from 400 to 1200 nm are plotted in Fig. 3d. The absorption spectra have five absorption peaks at 488, 522, 654, 792, and 975 nm, corresponding to the transitions of Er3+ ions from the ground state 4I15/2 to the 4F7/2, 2H11/2, 4F9/2, 4I9/2 and 4I11/2 excited states, respectively. Upon the introduction of In3+ ions the intensities of these absorption peaks are remarkably enhanced. The absorption cross-section (σabs) which is an indicator to evaluate the distribution of Er3+ ions are calculated using the equation \(\int_{{{\text{band}}}} {\sigma (\lambda )d\lambda = \sum\nolimits_{{{\text{band}}}} {{{\alpha (\lambda )} \mathord{\left/ {\vphantom {{\alpha (\lambda )} {Cd\lambda }}} \right. \kern-\nulldelimiterspace} {Cd\lambda }}} }\) [7, 37], where λ, α, and C are the absorption wavelength, absorption coefficient, and concentration of Er3+ ions, respectively. To reveal the intrinsic correlation between σabs and emission intensity, we plot σabs of 2H11/2 and the emission intensity of 550 nm band against the content In3+ content in the inset of Fig. 3d. The variation of the emission intensity with the In3+ content shows similar trend as that of σabs. Both σabs and emission intensity reach the maximum values at x = 1.75%. According to previous reports [5, 7, 38], the doped Er3+ ions have two different distribution sites, i.e., Er3+ isolated sites and cluster sites and In3+ can modify the distribution of Er3+ sites according to the Judd–Ofelt (J–O) theory [39, 40]. Since σabs of Er3+ isolated sites is larger than that of Er3+ cluster sites [41], it thus can be concluded that the isolated sites are minimum at x = 0 and reaches the maximum at x = 1.75%. Furthermore, considering the effect between the adjacent Er3+ ions, the CR processes more possible occur among Er3+ cluster sites [5, 42], \(({\text{i.e., CR1: }}{}^{4}{\text{S}}_{3/2} \to {}^{4}{\text{I}}_{9/2} + {}^{4}{\text{I}}_{15/2} \to {}^{4}{\text{I}}_{13/2} {\text{ and CR2: }}{}^{4}{\text{S}}_{3/2} \to {}^{4}{\text{I}}_{13/2} + {}^{4}{\text{I}}_{15/2} \to {}^{4}{\text{I}}_{11/2} )\) due to more small distance of Er3+ ions. The CR processes is no radiation relaxation process, the excited electrons decay to the low level without emission photons, influencing the efficiency of electrons radiative transition. In addition, the inherent ESA process happens among Er3+ isolated sites, which is the necessary condition for enhancing emission. Therefore, more Er3+ isolated sites generate, leading to more efficient ESA process and scarce CR process and the increase of both σabs and emission intensity, with the content of 0 ≤ x ≤ 1.75%. The x = 1.75% sample with the largest σabs and emission intensity should possess the maximal isolated Er3+ and the minimal cluster Er3+ sites. Notably, for x > 1.75%, the reason for the decrease of σabs and emission intensity may be due to the excess In3+ doping-induced formation of impurity level which have significant impact on the Er3+ energy level, reducing the efficiency of energy levels transition. According to the above analysis, all these interpretations consistent with the experimental results that In3+ can modify the Er3+ -sites distribution and Er3+ isolated sites, which is beneficial to the enhancement of upconversion luminescence intensity of B1−xCT:0.75%Er ceramics.
4 Conclusions
In3+-doped Ba0.85−xCa0.15TiO3:0.75%Er3+/xIn3+ upconversion photoluminescent ferroelectric ceramics have been prepared by the conventional solid-state method and the effects of In3+ content on the upconversion photoluminescent properties have been studied. Upon 980 nm excitation the 550 nm band upconversion emission intensity increases dramatically with increasing In3+ content and reaches the maximum at x = 1.75% mol, where the green emission intensity is unprecedentedly (~ 2000%) larger than that of the In3+ undoped Ba0.85Ca0.15TiO3:0.75%Er3+ ones. UV-VIS-NIR absorption spectra reveal that the In3+ doping increases Er3+ isolated sites and reduces Er3+ cluster sites, thereby favoring the excited state absorption process and reduces cross relaxation. As a result, the upconversion efficiency is greatly enhanced. Our results demonstrate that the nominal A-site In3+ doping is an effective approach to enhance the upconversion green emission efficiency of Er3+-doped perovskite ferroelectrics.
References
F. Auzel, Chem. Rev. 104, 139–174 (2004)
J. Ruan, Z. Yang, H. Zhang, J. Qiu, Z. Song, D. Zhou, Opt. Lett. 43, 3885–3888 (2018)
D. Gao, D. Wang, X. Zhang, X. Feng, H. Xin, S. Yun, D. Tian, J. Mater. Chem. C 6, 622–629 (2018)
W. Shi, Z. Zhang, J. Qin, Y. Zhang, Y. Liu, Y. Liu, H. Gao, Y. Mao, J. Alloy. Compd. 754, 124–130 (2018)
Y. Qian, R. Wang, B. Wang, B. Zhang, S. Gao, RSC. Adv. 4, 6652–6656 (2014)
L. Guo, J.M. Yan, Y.Y. Zhang, Z.X. Xu, M. Xu, L.H. Luo, F.F. Wang, Y.K. Liu, S. Dong, R.K. Zheng, J. Mater. Sci. 29, 9007–9015 (2018)
H. Zhou, Y.P. Huang, J.M. Yan, L.H. Luo, F.F. Wang, G.L. Yuan, R.K. Zheng, Mater. Lett. 188, 364–367 (2017)
Y. Fu, S. Gong, X. Liu, G. Xu, Z. Ren, X. Li, G. Han, J. Mater. Chem. C 3, 382–389 (2015)
T. Zheng, L. Luo, P. Du, A. Deng, W. Li, J. Eur. Ceram. Soc. 38, 575–583 (2018)
X. Wu, C. Fang, J. Lin, C. Liu, L. Luo, M. Lin, X. Zheng, C. Lin, Ceram. Int. 44, 4908–4914 (2018)
Z. Xu, J. Gong, A. Li, Y. Han, Y. Luo, J. Lumines. 135, 10–14 (2013)
Y. Zhang, J. Hao, C.L. Mak, X. Wei, Opt. Express 19, 1824–1829 (2011)
J. Wu, Z. Wu, W. Qian, Y. Jia, Y. Wang, H. Luo, Mater. Lett. 184, 131–133 (2016)
S. Wang, H. Zhou, X. Wang, A. Pan, J. Phys. Chem. Solids 98, 28–31 (2016)
Y. Chen, G.H. Chen, X.Y. Liu, T. Yang, J. Lumines. 195, 314–320 (2018)
Y. Chen, G. Chen, X. Liu, J. Xu, X. Zhou, T. Yang, C. Yuan, C. Zhou, Opt. Mater. 81, 78–83 (2018)
L. Li, K. Xu, Y.H. Wang, Z.F. Hu, H. Zhao, Opt. Mater. Express 6, 1022–1030 (2016)
L.L. Noto, S.S. Pitale, M.A. Gusowki, J.J. Terblans, O.M. Ntwaeaborwa, H.C. Swart, Powder Technol. 237, 141–146 (2013)
W. Zhang, Y.L. Liang, Z.F. Hu, Z.Y. Feng, M. Lun, X.P. Zhang, X. Sheng, Q. Liu, J. Luo, J. Nanosci. Nanotechnol. 16, 3517–3521 (2016)
H. Yu, Q. Huang, W. Cao, X. Zhang, J. Yu, Acta Chim. Sin. 71, 1639–1646 (2013)
H.R. Li, C.X. Chen, R.K. Zheng, J. Mater. Sci. 26, 3057–3063 (2015)
C.L. Freeman, J.A. Dawson, H.R. Chen, L.B. Ben, J.H. Harding, F.D. Morrison, D.C. Sinclair, A.R. West, Adv. Funct. Mater. 23, 3925–3928 (2013)
Y. Tsur, T.D. Dunbar, C.A. Randall, J. Electroceram. 7, 25–34 (2001)
H.D. Li, C.D. Feng, W.L. Yao, Mater. Lett. 58, 1194–1198 (2004)
Z.Q. Zhuang, M.P. Harmer, D.M. Smyth, R.E. Newnham, Mater. Res. Bull. 22, 1329 (1987)
Q. Yue, L. Luo, X. Jing, W. Li, J. Zhou, J. Alloy. Compd. 610, 276–280 (2014)
M. Wu, Y. Lu, Y. Li, J. Am. Ceram. Soc. 90, 3642–3645 (2007)
L. Luo, P. Du, W. Li, Q. Yue, J. Appl. Phys. 114, 124104 (2013)
N. Jeon, D. Rout, W. Kim, S.J.L. Kang, Appl. Phys. Lett. 98, 072901 (2011)
Q. Zuo, L. Luo, W. Li, F. Wang, J. Phys. D 49, 265303 (2016)
D.K. Khatua, A. Agarwal, A. Mshra, G.D. Adhikary, A. Senyshyn, R. Ranjan, J. Appl. Phys. 124, 104101 (2018)
E. Siegel, K.A. Muller, Phys. Rev. B 20, 3587–3596 (1979)
X. Wu, K.W. Kwok, F. Li, J. Alloy. Compd. 580, 88–92 (2013)
G. Chen, G. Somesfaleam, Y. Liu, Z. Zhang, Q. Sun, F. Wang, Phys. Rev. B 75, 195204 (2007)
P.V. dos Santos, E.A. Gouveia, M.T. de Araujo, A.S. Gouveia-Neto, A.S.B. Sombra, J.A. Medeiros, Neto, Appl. Phys. Lett. 74, 3607 (1999)
W. Li, Z. Xu, R. Chu, P. Fu, G. Zang, J. Alloy. Compd. 583, 305–308 (2004)
W.Q. Shi, M. Bass, M. Birnbaum, J. Opt. Soc. Am. B 7, 1456–1462 (1990)
Y. Qian, R. Wang, C. Xu, W. Wu, X. Wu, C. Yang, J. Alloy. Compd. 527, 152–156 (2012)
M.D. Shinn, W.A. Sibley, M.G. Drexhage, R.N. Brown, Phys. Rev. B 27, 6635 (1983)
M.P. Hehlen, N.J. Cockroft, T.R. Gosnell, A.J. Bruce, Phys. Rev. B 56, 9302 (1997)
D.M. Gill, L. McCaughan, J.C. Wright, Phys. Rev. B 53, 2334 (1996)
Y. Wang, J. Ohwaki, Appl. Phys. Lett. 63, 3268–3270 (1993)
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 51572278).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, TW., Xu, ZX., Zhang, YY. et al. Huge enhancement of upconversion photoluminescence in In3+ doped Ba0.85−xCa0.15TiO3:0.75%Er3+/xIn3+ lead-free ferroelectric ceramics. J Mater Sci: Mater Electron 30, 4455–4461 (2019). https://doi.org/10.1007/s10854-019-00734-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-019-00734-1