
Abstract
The structural, optical, and dielectric properties of LaFe1−xMoxO3 (x = 0.0, 0.5) synthesized by the sol–gel method were investigated by X-ray diffraction (XRD), Raman scattering, and impedance spectroscopy. XRD analysis revealed that the x = 0.5 compound is single-phase and orthorhombic, with space group Pbnm, similar to the parent x = 0.0 compound but with larger lattice parameters and/or a larger unit-cell volume. The Raman scattering spectra revealed local lattice distortions in the x = 0.5 compound and spin–phonon coupling related to the magnetic transition (Néel) temperature. The impedance data were well fitted with an R(R1-CPE1)//(R2-CPE2) equivalent electrical circuit, demonstrating the contributions of both grains and grain boundaries. The temperature (300–500 K) and frequency (100 Hz–1 MHz) dependences of the dielectric properties showed that Mo substitution adversely affected the dielectric parameters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Perovskite ceramics are simple ionic compounds with general chemical formula ABO3, where A is usually a large cation and B is generally a medium-sized cation. They have been intensively studied because of their unique physical properties, which include unusual dielectric, piezoelectric, ferroelectric, magnetic ordering, multiferroic, electronic conductivity, thermal, and optical properties. Furthermore, these materials have received broad attention because of their potential practical applications. Materials with high oxide ionic conductivity, high electronic conductivity, or a mixture of both are required for chemical sensors, etc. Additionally, the substitution of cations at A and/or B sites results in new and unexpected responses compared with those of the parent ABO3 [1, 2]. In particular, several complex perovskites of general formula \(~A(B_{{0.5}}^{'}B_{{0.5}}^{{''}}){{\text{O}}_3}\) are more attractive for applications such as sensors, electrode materials, detectors, and actuators [1, 3]. Several researchers have reported the structural and electrical properties of \({\text{Sr}}({\text{B}}{{\text{i}}_{0.5}}{\text{N}}{{\text{b}}_{0.5}}){{\text{O}}_3}\) [1], which exhibit an orthorhombic perovskite crystal structure, low conduction-process activation energies, and a low tangent loss. This material thus has potential applications in electronics devices.
Lanthanum orthoferrite, LaFeO3, is a perovskite-structured material with potential applications in advanced technologies, especially as a sensor material and an electrode material in solid oxide fuel cells (SOFCs) [4]. Previous studies have revealed that LaFeO3 exhibits multiferroic characteristics at room temperature [5, 6]. LaFeO3 is an antiferromagnetic material with a Néel temperature (TN) of approximately 740 K and undergoes a phase transition to a rhombohedral structure at approximately 987 °C [4]. LaFeO3 synthesized via a polycondensation reaction showed high sensitivity and stability toward formaldehyde gas. It has potential applications as a formaldehyde gas sensor, as reported by Zhang et al. [7]. The substitution of other ions into the La and/or Fe sites can improve the physical properties of LaFeO3, resulting in, for example, a stable structure with high thermal stability, a high dielectric constant, and low dielectric losses [2]. In La0.8Ca0.2−xPbxFeO3, in which Ca and Pb have been doped into La sites, the best sensitivity toward ethanol was observed for the composition with x = 0.1 at.% in a study that focused on electrical characterization of the changes in resistance [8]. Bhargav et al. have reported that (La, Pb)FeO3 with Pb doped at 0.2 at.% into La sites exhibits colossal dielectric constant behavior [9]. Tahir Murtaza et al. also have reported that Mo-doped BiFe1−xMoxO3 (x = 0.0, 0.6) exhibits the changes in structure, increased the grain size, reduction in leakage current density (enhancement in polarization), and enhanced the ferroelectricity (by seen the increased in remnant and maximum polarization) [10]. By contrast, research into LaFeO3 with Mo doped into Fe sites has not yet been reported. The partial substitution of Mo into the Fe sites of LaFeO3, resulting in LaFe1−xMoxO3, is expected to exhibit physical properties that differ from those of the parent (x = 0.0) compound. The starting point to explain the electrical properties of LaFe1−xMoxO3 is the valence of the metal ions. In LaFeO3 and LaMoO3, Fe and Mo ions are both in a formal 3+ valence state. When they mix to form LaFe1−xMoxO3, the charges of the metal ions are adjusted, which will affect the structural and physical properties of this compound.
In the present study, we focus on partial Mo substitution into the Fe sites of LaFe1−xMoxO3 and compare our results with those for the parent (x = 0.0) compound. LaFe1−xMoxO3 with x = 0.0 and 0.5 were synthesized by a combined sol–gel and sintering method. The structural and physical properties of these materials were investigated.
2 Experimental details
The crystalline LaFe1−xMoxO3 (x = 0.0, 0.5) powders were prepared by the sol–gel method using lanthanum oxide, ferrite nitrate nonahydrate, molybdenum(VI) oxide, and citric acid monohydrate as raw materials (all analytical grade). All of the precursors were solved and stirred to form a gel, which was then heated at 200 °C to form a brown powder. The as-prepared powders were subsequently calcined at 900 °C for 6 h. The samples were then pressed into pellets with a diameter of 12 mm at a pressure of 10 kN for 5 min and sintered at 1300 °C for 1 h.
The structural phases were identified by X-ray diffraction (XRD) with Cu Kα radiation in the 2θ range from 20° to 80° with a step size of 0.02°. Rietveld refinement of the diffraction pattern was carried out using the FullProf 2k software. Raman scattering measurements were recorded in the range from 70 to 1600 cm−1 with an excitation wavelength of 532 nm. The samples were placed in a LINKAM heating stage system, allowing measurements in the temperature range from 300 to 800 K. The presence of phase transition of the samples was measured by differential scanning calorimetry (DSC) at heating rate of 10 °C/min from 150 to 475 °C and encapsulated in Al crucibles under nitrogen atmosphere. Dielectric properties of LaFe1−xMoxO3 (x = 0.0, 0.5) were characterized using a FLUKE PM6306 RLC meter in the frequency range between 100 Hz and 1 MHz and in the temperature range from 300 to 500 K. Dielectric measurements for this material were analyzed via the parallel-plate method using impedance data. The morphology and microstructure of the samples were examined by scanning electron microscopy (SEM) (Merk. FEI QUANTA 650).
3 Results and discussion
3.1 X-ray diffraction
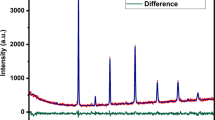
Figure 1 shows the XRD patterns of the LaFe1−xMoxO3 (x = 0.0, 0.5) sintered at 1300 °C in air. The structural analysis was carried out using the FullProf 2k software. The analysis confirmed that the structure for the x = 0.0 composition is an orthorhombic crystal structure with space group Pbnm. The obtained fitting parameters for all of the samples are shown in Table 1.

XRD patterns of LaFe1−xMoxO3 (x = 0.0, 0.5) after sintering process. The black cross symbols are the experimental data. The red lines are refinement results. The blue lines are the difference between the experimental data and refinement results
The substitution of Mo4+ ions into Fe sites was expected to result in different lattice parameters between the x = 0.0 and x = 0.5 compositions. The lattice parameters of the x = 0.5 composition are slightly larger than those of the x = 0.0 composition because the ionic radius of Fe2+ (77 pm) is smaller than that of Mo4+ (79 pm), although the difference is not substantial. According to XRD refinement results (Table 1) for the x = 0.0 and x = 0.50 compositions, the occupancy factor indicates that Mo ions are substituted into Fe sites in the crystal lattice. The absence of secondary phases after sintering indicates that the substitution of Fe2+ ions with Mo4+ ions does not achieve the solvability limit. Certainly, the degree of host cation substitution by other ions depends on their radius [11]. For example, the Mo solvability in LaFeO3 is caused by small difference between the ionic radii of Fe3+ (63 pm) and Mo6+ (73 pm) ions. The replacement of the Fe3+ ions by the Mo6+ ions induces the formation of Fe2+ and Mo4+ ions in the crystal lattice, resulting in electric charge compensation [3].
Using the VESTA software, we calculated the geometrical parameters characterizing the crystal structure of both samples; the results are given in Table 2. The tolerance factor \((t)\) can be calculated to ensure the stability of the crystal structure of perovskite \(ABB\text{'}{O_3}\).
where rA, rB, and rB′ are the radii of the A, B, and B′ cations, respectively, and rO is the radius of an oxide ion [11].
For 0.75 < t < 1.0, the perovskite structure is formed with different distortions, such as orthorhombic or rhombohedral, but remains stable [11]. Upon Mo substitution, the orthorhombic deformation, \(D\), increases, whereas t decreases, indicating that the lattice becomes more distorted. The tolerance factor being smaller than unity (t < 1.0) also means that tilted FeO6 octahedra are present. The two Fe–O–Fe angles \({\theta _1}\) and \({\theta _2}\) are not 180° because of the tilt of FeO6 octahedra in the pseudo-cubic [1 1 1] direction [6]. The average tilt angle, \(\varphi\), slightly increases accordingly, indicating an increase in the distortion of the FeO6 octahedra.
3.2 Raman scattering spectra
Figure 2 shows the room-temperature Raman scattering spectra of LaFe1−xMoxO3 (x = 0.0, 0.5). The spectra are composed of several phonon modes and a broad photoluminescence background. We fitted these phonon peaks using a standard Lorentzian profile. A broad luminescence background could be due to the defects in the samples. The frequencies of some specific phonon modes shown in Fig. 2 are listed in Table 3. Our result for the x = 0.0 composition is consistent with a previously reported Raman scattering spectrum of LaFeO3 [12].

Raman scattering spectra of LaFe1−xMoxO3 (x = 0.0, 0.5) at room temperature. The dashed lines are the best fit using the Lorentzian model
According to factor group analysis, LaFeO3 has an orthorhombic perovskite structure (space group Pbnm) containing four formula units per primitive cell. Among the total 60 Γ-point normal modes, 24 (7Ag + 7B1g + 5B2g + 5B3g) are Raman-active, 25 (7B1u + 9B2u + 9B3u) are infrared-active, 3 (B1u + B2u + B3u) are acoustic modes, and 8 (8Au) are silent modes. As confirmed in previous reports [13,14,15], the modes below 200 cm−1 (ω1, ω2, and ω3) are related to vibrations of the La-site cations with Ag symmetry. The mode at approximately 267 cm−1 (ω4) is attributed to the tilt vibrations of FeO6 octahedra with Ag symmetry. The mode at approximately 297 cm−1 (ω5) is assigned to the mixed vibrations of La-site site and O1-atoms with B1g symmetry. The mode at approximately 414 cm−1 (ω6) is associated with the bending vibrations of FeO6 octahedra with Ag symmetry. The modes at approximately 436 cm−1 (ω7) and 473 cm−1 (ω8) are due to the asymmetric stretching vibrations associated with Jahn–Teller distortions with Ag symmetry. The mode at approximately 629 cm−1 (ω9) is assigned to the symmetric stretching vibrations of FeO6 octahedra with B1g symmetry. Additional phonon modes observed above 700 cm−1 should be ascribed to multiphonon processes [16, 17].
In the case of the x = 0.5 composition, the Raman scattering intensity of all phonon modes is reduced and their linewidth broadens, reflecting an increase of lattice disorder. This is consistent with the observation of an increase in the distortion of the FeO6 octahedra by X-ray diffraction experiments. Indeed, Mo substitution modifies the lattice constants, causing a change in the Jahn–Teller distortions [18, 19]. Notably, the appearance of new multiphonons in the range of 750–1000 cm−1 is certainly related to Mo atoms, which occupy B-site octahedral positions. They are possibly activated by the local electron–phonon coupling according to the Franck–Condon picture following Fe–Mo charge transfer [16].
We now turn to address the temperature-dependent Raman scattering spectra of LaFe1−xMoxO3 (x = 0.0, 0.5), shown in Fig. 3. No substantive change in the Raman shift is observed with increasing temperature for either sample, indicating that the crystal structure of both samples remains unchanged. Any change in a material’s structure or physical properties can alter phonon characteristics such as the frequency (Raman shift), linewidth, and intensity of the Raman peaks [15]. The spectra for both samples show that increasing temperature causes the shifts of the Raman modes to lower frequency, broadening of peaks, and a decrease of peak intensity.

Raman scattering spectra of LaFe1−xMoxO3 (x = 0.0, 0.5) compounds as a function of temperature
Figure 4 shows the temperature-dependent phonon parameters (frequency, linewidth, and normalized intensity) of the Jahn–Teller and stretching modes of FeO6 octahedra in the LaFe1−xMoxO3 (x = 0.0, 0.5) compounds. Increasing temperature clearly causes a shift of Raman modes to lower frequency, linewidth broadening, and diminished intensity. The phonon modes tend to exhibit anomalies of the phonon parameters at approximately 700 K for x = 0.0 and 675 K for x = 0.5. These anomalies indicate the presence of spin–phonon interactions related to the magnetic transition temperature (Néel temperature) [20,21,22]. The Mo substitution causes the Néel temperature to shift to lower temperatures.

Temperature dependences of the Raman phonon parameters of the Jahn–Teller and stretching modes of FeO6 octahedra in the LaFe1−xMoxO3 (x = 0.0, 0.5) compounds. Vertical dashed lines denote transition temperatures
3.3 Differential scanning calorimetry analysis
Figure 5 shows the differential scanning calorimetry (DSC) thermogram of LaFe1−xMoxO3 (x = 0.0, 0.5). The thermogram confirms the order–disorder of transition temperatures as the heat-flow peak temperatures. In this case, the peak temperature corresponds to a phase transition to an antiferromagnetic phase (i.e., TN) [23, 24]. The TN occurs at approximately 418 °C (691 K) for x = 0.0, which is similar to the previously reported value of ~ 463 °C (736 K) [4]. The difference of TN for LaFeO3 compared with the reported one might be related with the crystallite/grain size, which is strongly influenced by the chemical-solid reaction during the synthesis process [25, 26]. The presence of an antiferromagnetic phase was also confirmed by the results of high-temperature Raman phonon parameter analysis (Fig. 4).

DSC thermogram of LaFe1−xMoxO3 (x = 0.0, 0.5) compounds
The superexchange interaction is known to depend on the valence of magnetic ions; i.e., they must have the same valence and filled d-shells (partially filled shells result in the strongest interaction), the interaction must occur in the eg levels, and the octahedral bond angle should approach 180° (180° results in the greatest stabilization) [18, 19]. On the basis of the structural analysis (Tables 1, 2), Mo substitution clearly increases the deviation of the superexchange Fe–O–Fe bond angle from 180° and the Mo ions occupy the Fe-sites in the crystal with an ionic state Mo4+\({{\text{d}}_2}{\text{t}}_{{2{\text{g}}}}^{2}{\text{e}}_{{\text{g}}}^{0}\). Consequently, the superexchange interaction decreases, as indicated by the decrease of TN. The change in the magnetic ion states from Fe3+\({{\text{d}}_5}{\text{t}}_{{2{\text{g}}}}^{3}{\text{e}}_{{\text{g}}}^{2}\) (x = 0.0) to Fe2+\(~{{\text{d}}_6}{\text{t}}_{{2{\text{g}}}}^{4}{\text{e}}_{{\text{g}}}^{2}\) (x = 0.5) does not reduce the superexchange interaction (i.e., the interaction occurs in the eg levels for Fe3+ and Fe2+ magnetic ions).
3.4 Dielectrical properties
We used impedance spectroscopy to characterize the relaxation and conduction processes and to identify the charge carriers in the conduction and/or relaxation processes. The magnitude of the impedance (Z) and phase angle was measured over the frequency range from 100 Hz to 1 MHz and over the temperature range from 27 to 227 °C. The imaginary part of the impedance was calculated for all temperatures in the investigated range.
Figure 6a shows the complex impedance spectrum (Nyquist plot) of LaFe1−xMoxO3 (x = 0.5) measured from 27 to 227 °C (x = 0.0 not shown in this work). With increasing temperature, the semicircle in the complex impedance spectrum decreases in amplitude, indicating increasing conductivity of the sample [1]. The impedance data were fitted to deduce the equivalent circuit. The best fit was achieved with two parallel combinations of resistance (R) and a constant phase element (CPE): R(R1-CPE1)//(R2-CPE2). This equivalent circuit is consistent with the observation of two depressed semicircle arcs in the measured frequency range from 100 Hz to 1 MHz. The semicircle formed at high frequency is dominated by bulk (grain) contributions, whereas the semicircle at low frequency corresponds to grain-boundary contributions.

a Nyquist plot and b Bode plot of \(Z^{\prime\prime}(f)\) for LaFe1−xMoxO3 (x = 0.5) at 300 K, 323 K, 423 K, 473 K, and 498 K
The conductivity, which comprises bulk (grain) \(({\sigma _g})\) and grain boundary \(({\sigma _{gb}})\) contributions, was calculated using Eqs. (2) and (3) [27]:
where \(t\) is thickness, \(A~\)is surface area of the samples, and \({R_g}\) and \({R_{gb}}\) are resistivity contributed by grain and grain boundary, respectively. Table 4 shows the conductivity values for both samples. For the x = 0.0 composition, the \({\sigma _g}\) is larger than \({\sigma _{gb}}\) for T ≤ 423 K and \({\sigma _g}\) is smaller than \({\sigma _{gb}}\) for T > 423 K, indicating that the electrical conductivity is dominated by grain responses at T ≤ 423 K and by grain-boundary responses at T > 423 K. By contrast, for the x = 0.5 sample, \({\sigma _g}\) is smaller than \({\sigma _{gb}}\), suggesting that grain-boundary contributions dominate the electrical conductivity.
Calculating the activation energy (Ea) of the charge carriers in the conduction process requires calculating the conductivity σg and σgb for each temperature. The Ea obtained from dc transport measurements was calculated using the Arrhenius relation (Eq. 4) [28]:
where \({k_{\text{B}}}\) is the Boltzmann constant.
The value of Egb indicates the concentration of ionic vacancies, in this case oxygen vacancies, in samples, which is related to ionic conductivity [29, 30]. However, the crystallographic analysis (Table 1) indicates that the valences of the two samples are balanced by ions in the lattice. This observation suggests that another factor must be responsible for the ionic conductivity; specifically, oxygen in the lattice moves to other sites (defects), introducing oxygen vacancies, possibly during the preparation process [29, 30]. The Egb of the x = 0.0 sample is larger than that of the x = 0.5 sample, indicating that the Mo substitution increases oxygen-ion migration, thereby increasing the oxygen vacancy concentration in the lattice, which leads to an increase of the oxygen-ion conductivity and a decrease of Egb. For grain contributions, the Eg is associated with the presence of electronic charge carriers, i.e., electron and/or hole conduction, in the crystal lattice. The Eg of the x = 0.0 sample is smaller than that of the x = 0.5 sample, indicating that Mo substitution increases the energy barrier blocking the movements of the space charge in the lattice.
Figure 6b shows the imaginary parts of impedance as a function of frequency at different temperatures for the x = 0.5 sample. The imaginary component (Z″) of impedance increases gradually until a maximum point and then decreases, indicating a relaxation phenomenon. The Z″ peak shifts toward higher frequencies with increasing temperature, indicating an increase of the relaxation time and suggesting the occurrence of thermal processes in this material. Such behavior can originate from the existence of immobile species at lower temperatures and defects at higher temperatures [28].
To understand the dielectric properties of LaFe1−xMoxO3 in the temperature range 300–500 K, we investigated the variation of the dielectric constants as a function of frequency in the frequency range from 100 Hz to 1 MHz; the results are given in Fig. 7. For both compounds, the dielectric constant clearly decreases with increasing frequency. The dielectric constant increases at lower frequencies and is independent at higher frequencies. The high dielectric constant at lower frequencies can be caused by migration of oxygen vacancies as an ionic charge carrier at the interface or grain boundary (defect) [31]. The dielectric constant also increases with increasing of temperature. This behavior is explained by the inability of electric dipoles to orient themselves with the applied electric field. An increase of temperature promotes dipole orientation. The increasing orientational polarization leads to an increase of the dielectric constant with increasing temperature [31]. The Mo substitution lowers the dielectric constant.

Dielectric constant of LaFe1−xMoxO3 with ax = 0.0 and bx = 0.5 at different temperatures
As Mallah [31] reported for the dielectric relaxation phenomenon for perovskite compounds, the dielectric loss \((\varepsilon ^{\prime\prime})\) can be caused by a conduction loss, dipole loss, or a vibrational loss. Figure 8 shows the \(\varepsilon ^{\prime\prime}\) of LaFe1−xMoxO3 (x = 0.0, 0.5) as a function of frequency at different temperatures. The inset of Fig. 8a describes the loss dielectric of x = 0.0 sample at 300 K and 323 K. The peak indicates the temperature dependence on dielectric relaxation phenomenon occurring in the system. The magnitude of \(\varepsilon ^{\prime\prime}\) increases with increasing temperature. Similar to the trend for \(\varepsilon ^{\prime}\) (Fig. 7), the \(\varepsilon ^{\prime\prime}\) decreases with increasing frequency, which is attributed to the migration of oxygen ions being the main cause of the dielectric loss at low frequencies. The high value of \(\varepsilon ^{\prime\prime}\) at low frequencies can arise from the contribution of ionic charge-carrier jumping and conduction losses due to ion migration, which induces ionic polarization. By contrast, in the higher frequency range, ion vibrations may be the only cause of decreasing dielectric loss.

Dielectric loss of LaFe1−xMoxO3 with ax = 0.0 and bx = 0.5 at different temperatures. The inset of a describes the loss dielectric of x = 0.0 at 300 K and 323 K
The obtained results for \(\varepsilon ^{\prime\prime}\) can be analyzed according to the hopping theory of charge carriers over a potential barrier between charge defects [31]. On the basis of this model, the \(\varepsilon ^{\prime\prime}\)can be expressed by the following relation:
where
where \(R\) is constant, \(m~\) is a frequency power parameter, and \({W_m}\) is the maximum barrier height. Using Eqs. (5) and (6), we obtained the frequency power parameter \(m\) and maximum barrier height. Figure 9 shows the temperature dependence of the power parameter, which decreases linearly with increasing temperature. Mo substitution into Fe sites clearly causes an enhancement of the \({W_m}\) value. Thus, overcoming the trapping of hopping charge carriers trapped in localized sites would be more difficult.

Temperature dependence of power parameter \(m\) for LaFe1−xMoxO3 with ax = 0.0 and bx = 0.5
3.5 SEM images
Figure 10 illustrates the SEM photographs and energy dispersive analysis of X-ray EDAX spectra of LaFe1−xMoxO3 (x = 0.0, 0.5) compounds. The SEM images reveal the presence of grain distribution connecting with each other. Moreover, the EDAX spectra disclose the presence of all elemental compositions for each samples, indicating the absence of any integrated element during sintering process. The difference in the shape and size of grain indicates the existence of polycrystalline characteristics throughout the samples. The average grain size is estimated to be about 2.1 and 0.34 µm for x = 0.0 and x = 0.5, respectively. This indicates that the distribution of Mo-ions at grain boundaries inhibits the grain growth of its parent, LaFeO3.

SEM and EDAX analysis spectra of LaFe1−xMoxO3 (x = 0.0, 0.5) compounds. The EDAX of x = 0.5 describes the observation of Mo-accumulation in the grain boundary labeled by “+” (SEM-inset)
4 Conclusion
We synthesized LaFe1−xMoxO3 (x = 0.0, 0.5) by the sol–gel method and investigated their structural, optical, and dielectric properties. XRD analysis confirmed that x = 0.5 has an orthorhombic perovskite crystal structure with space group Pbnm, similar to the parent x = 0.0 compound but with larger lattice parameters and/or a larger unit-cell volume. Raman scattering analysis revealed local lattice distortions and a shift of the TN to a lower temperature at x = 0.5, as also confirmed by DSC. The impedance analysis revealed the presence of both a grain and a grain-boundary response in the conduction mechanism. The grain responses reveal that Mo substitution causes a higher Eg; that is, Mo substitution increases the energy barrier that blocks the movements of space charges in the lattice. By contrast, the grain-boundary responses show a decreasing Egb, indicating increases of both oxygen ionic migration and the oxygen vacancy concentration in the lattice, which led to an increase of the oxygen ionic conductivity. The SEM images revealed that Mo-ions reduced the grain size. The space charges and oxygen ions (vacancies and migration) are related to the electronic and ionic conductivity, respectively. The dielectric properties of LaFe1−xMoxO3 (x = 0.0, 0.5) confirmed the decrease of the dielectric parameters due to the Mo substitution.
References
B.C. Sutar, R.N.P. Choudhary, P.R. Das, Ceram. Int. 40, 7791–7798 (2014)
C. Xueying, L. Shengli, Z. Xinde, Mater. Lett. 130, 267–270 (2014)
S. Phokha, S. Hunpratup, S. Pinitsoontorn, B. Putasaeng, S. Rujirawat, S. Maensiri, Mater. Res. Bull. 67, 118–125 (2015)
R. Köferstein, L. Jäger, S.G. Ebbinghaus, Solid State Ion. 249–250, 1–5 (2013)
S. Acharya, J. Mondal, S. Ghosh, S.K. Roy, P.K. Chakrabarti, Mater. Lett. 64, 415–418 (2010)
Z. Zhou, L. Guo, H. Yang, Q. Liu, F. Ye, J. Alloys Compd. 583, 21–31 (2014)
H. Zhang, P. Song, D. Han, Q. Wang, Physica E 63, 21–26 (2014)
A. Benali, S. Azizi, M. Bejar, E. Dhahri, M.F.P. Graça, Ceram. Int. 40, 14367–14373 (2014)
K.K. Bhargav, S. Ram, S.B. Majumder, J. Alloys Compd. 638, 334–343 (2015)
T. Murtaza, I.A. Salmani, J. Ali, M.S. Khan, J. Supercond. Novel Magn. (2018). https://doi.org/10.1007/s10948-017-4443-4
C. Doroftei, P.D. Popa, F. Iacomi, L. Leontie, Sens. Actuators B 191, 239–245 (2014)
A.S. Mahapatra, A. Mitra, A. Mallick, M. Ghosh, P.K. Chakrabarti, Mater. Lett. 169, 160–163 (2016)
M.L. Sanjuan, V.M. Orera, R.I. Rerino, J. Blasco, J. Phys.: Condens. Matter 10, 11687–11702 (1998)
L.M. Carron, A. de Andres, M.J.M. Lope, M.T. Casais, J.A. Alonso, J. Alloys Compd. 323–324, 494–497 (2001)
T. Runka, M. Berkowski, J. Mater. Sci. 47, 5393–5401 (2012)
J. Andreasson, J. Holmlund, R. Rauer, M. Kall, L. Borjesson, C.S. Knee, A.K. Eriksson, S.G. Eriksson, M. Rubhausen, R.P. Chaudhury, Phys. Rev. B 78, 235103–235113 (2008)
L.M. Carron, A. de Andres, M.J.M. Lope, M.T. Casais, J.A. Alonso, Phys. Rev. B 66, 174303 (2002)
R.J.D. Tilley, Perovskite: Structure-Property Relationships (Wiley, New York, 2016)
R.P. Liferovich, R.H. Mitchell, J. Solid State Chem. 177, 2188–2197 (2004)
P.V. Coutinho, F. Cunha, P. Barrozo, Solid State Commun. 252, 59–63 (2017)
R. Rawat, D.M. Phase, R.J. Choudhary, J. Magn. Magn. Mater. 441, 398–403 (2017)
A. Panchwanee, V.R. Reddy, A. Gupta, V.G. Sathe Mater. Chem. Phys. 196, 205–212 (2017)
J. Wei, R. Haumont, R. Jarrier, P. Berthet, B. Dkhil, J. Appl. Phys. 111, 114106 (2012)
F.A. Mir, Int. J. Thermophys. 36, 1654–1660 (2015)
H. Das, A. Inukai, N. Debnath, T. Kawaguchi, N. Sakamoto, S.M. Hoque, H. Aono, K. Shinozaki, H. Suzuki, N. Wakiya, J. Phys. Chem. Solids 112, 179–184 (2018)
R. Tlili, M. Bejar, E. Dhahri, A. Zaoui, E.K. Hlil, L. Bessais, Polyhedron 121, 19–24 (2017)
P. Dhak, D. Dhak, M. Das, Pramanik, P, J. Mater. Sci. Mater. Electron. 22, 1750–1760 (2011)
A. Benali, M. Bejar, E. Dhahri, M.F.P. Graça, L.C. Costa, J. Alloys Compd. 653, 506–512 (2015)
K. Devi Chandrasekhar, S. Mallesh, J. Krishna Murthy, A.K. Das, A. Venimadhav, Physica B 448, 304–311 (2014)
Q. Ke, X. Lou, Y. Wang, J. Wang, Phys. Rev. B 82, 024102 (2010)
H.M. El-Mallah, Acta Phys. Pol. A 122, 174–179 (2012)
Acknowledgements
H.L.L. thanks financial support from the Ministry of Science and Technology of Republic of China under Grants No. MOST 105-2112-M-003-013-MY3. D Triyono thanks financial support from the Ministry of Technology Research and Higher Education of Republic of Indonesia under Grants No. 537/UN2.R3.1/HKP05.00/2018, 2nd year.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Triyono, D., Laysandra, H. & Liu, H.L. Structural, optical, and dielectric studies of LaFe1−xMoxO3 (x = 0.0, 0.5) perovskite materials. J Mater Sci: Mater Electron 30, 2512–2522 (2019). https://doi.org/10.1007/s10854-018-0525-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-018-0525-8