
Abstract
The size and shape of functional micro/nanomaterials have crucial influence on their physical and chemical properties. The monoclinic Y4O(OH)9NO3 with nanosheets, submicrospheres, and microrods have been successfully prepared by a non-aqueous solvothermal method. The precursors Y4O(OH)9NO3 decompose into cubic Y2O3:Eu3+ phosphors after sintering at 800 °C for 2 h. The morphologies and sizes of Y2O3:Eu3+ particles can be easily controlled by changing the growth rates of various crystallographic facets during solvothermal process. With the increment of the rare-earth nitrates concentration, the morphologies transform from 2D nanosheets to sub-microspheres, and eventually to microrods. The bright red emission, owing to Eu3+ f-orbital transitions, can be easily observed under ultraviolet excitation. Furthermore, the dependence of Y2O3:Eu3+ luminescence performances on different morphologies are discussed in the present work.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Luminescence materials have attracted great interest for decades because of their important significance in the daily life of human beings [1,2,3,4]. Much work has been done to satisfy the needs of bright light, tunable color, and high efficiency [5,6,7]. It is well-known that the size and shape affect the defects and dopant concentrations on the surface, which leads to nonradiative transitions and luminescence quenching. Therefore, the controlled synthesis of luminescence materials with desired sizes and shapes has received significant attention due to the potential for luminescence enhancement [8,9,10]. When luminescence materials are combined with therapeutic agents, multifunctional probes are synthesized for imaging-guided and tumor-targeted therapies [11,12,13]. Yttrium oxide is an outstanding host material because of its large band gap energy, good thermal property and high chemical stability, which has been extensively applied in advanced ceramics, dye sensitized solar cells, cathode-ray tubes, biomarkers and fluorescent lamps [14,15,16,17,18]. Y2O3:Eu3+, acting as dopant, improves the superconducting transition temperature of MgB2 bulk by strengthening the electron–phonon interaction [19, 20]. Y2O3 is easily replaced with various lanthanide ions with a valence of + 3 and used as a host for both down- and up-conversion luminescence [6, 9, 18]. In addition, various colors have been realized by proper selection and combination of lanthanide dopants. Y2O3:Eu3+ phosphors have been synthesized by many methods such as sol–gel, coprecipitation, hydro-/solvothermal, spray pyrolysis, combustion and microwave hydrothermal method [1, 16, 21, 22]. The hydro-/solvothermal method is a facile and efficient wet-chemistry method which has been successfully used to synthesize a large amount of materials with high purity and good uniformity [23,24,25,26]. The morphologies can be controlled by adjusting the critical factors including temperature, reaction time, additives, solvents, and so on. Nowadays, various morphologies of Y2O3:Eu3+ phosphors, including nanosheets, nanorods, nanotubes, flower-like and hierarchical structures have been prepared by hydro-/solvothermal method [27,28,29,30].
The evolution and design of morphologies have been deeply researched because of the complicated and unclear mechanisms [31,32,33,34]. The two-dimensional (2D) Y2O3:Eu3+ nanosheets could also be rolled or curled into the 1D nanotubes which then grew longer and thicker driven by the attached nanosheets [28]. The ultrathin, crystalline Y2O3 nanosheets can be assembled into a high surface area and low-density 3D network by a simple centrifugation-induced gelation method [22]. The 3D flower-like La2O3:Eu3+ composed of 1D nanorods was synthesized through a sequence of nucleation, crystal splitting, and self-assembly process assisted by oriented attachment [35]. The 3-dimensional flower-like Y2O3:Eu3+ nanoarchitectures assembled with nanosheets were synthesized by simple hydrothermal reaction without employing templates or matrix for self-assembly [27]. Thus, the evolution mechanism of different morphologies, like 1D rods/tubes, 2D sheets and 3D spheres, are worth studying.
This study reports the Y2O3:Eu3+ nanosheets, microspheres, and microrods which can be easily synthesized through a non-aqueous solvothermal method and subsequent heat treatment without any assistance of the surfactants and templates. The experiment results showed that the morphologies of the products could be greatly affected by the concentration of yttrium nitrate and volume ratio of benzyl alcohol and 1-octylamine. We investigated the photoluminescence properties of the as-obtained Y2O3:Eu3+ and discussed the relationship between luminescence intensity and morphologies. This new strategy can also be applicable in the preparation of other rare earth oxides whose chemical and physical properties are similar to yttrium oxide.
2 Experimental
2.1 Materials and methods
Yttrium oxide [Y2O3; Sinopharm Chemical Reagent Co., Ltd. (99.99%)], europium oxide [Eu2O3; Sinopharm Chemical Reagent Co., Ltd. (99.99%)], benzyl alcohol [C6H5CH2OH; Tianjin TIANLI Chemical Reagent Ltd. (AR)], 1-octylamine [C8H17NH2; Shanghai Kefeng Chemical Reagent Co., Ltd. (99%)], and nitric acid [HNO3; Sichuan Xilong Chemical Industry Co., Ltd. (65–68%)] were used in this study. All chemicals were used as received without further purification.
The following synthesis was performed after minor modifications to Ref. [21]. In a typical synthesis, 0.153 g of Y2O3 and 0.012 g of Eu2O3 (molar ratio Y:Eu = 0.95:0.05) were dissolved in 3 mL nitric acid heated at 70 °C. Rare earth nitrates were prepared by evaporating the superfluous nitric acid under constant heating. Afterwards, the rare earth nitrates were mixed with 12 mL of benzyl alcohol under magnetic stirring. After completely dissolving, 3 mL of 1-octylamine were added into the solution drop-by-drop with constant stirring. Furthermore, the mixture was stirred for another 30 min and transferred into a 25 mL teflon-lined autoclave for 24 h of solvothermal treatment at 160 °C. When the autoclave was cooled to room temperature naturally, the precursors were collected by centrifugation, washed several times with ethanol and dried at 60 °C in air. The as-synthesized powders were annealed at 800 °C for 2 h in air with a heating rate of 5 °C/min.
The benzyl alcohol/1-octylamine volume ratio and the concentration of Y1.9Eu0.1(NO3)3 were applied to control the morphology and size of Y2O3:Eu3+. The samples prepared at benzyl alcohol/1-octylamine volume ratio of 12/3, 9/6, 6/9 and 3/12 were labelled as S1, S2, S3 and S4. The samples prepared with 0.0765 and 0.306 g Y2O3 (Y:Eu kept at 0.95:0.05) were labelled as S5 and S6, respectively. The calcined samples for S1‒S6 were denoted as C1‒C6.
2.2 Characterizations
X-ray powder diffraction patterns were recorded by a X’Pert Pro (PANalytical B.V., The Netherlands) powder diffractometer with Cu Kα radiation. The FESEM images were obtained by a Quanta 600 FEG (FEI America) field emission scanning electron microscope equipped with an energy-dispersive spectrometer (EDS). TEM images were obtained by a Tecnai G2 F20 S-TWIN (FEI America) transmission electron microscope. Fourier transform infrared spectroscopy (FT-IR) spectra were measured by a JASCO FT/IR-470 plus infrared spectrophotometer with the KBr pellet technique. Thermogravimetric and differential scanning calorimetric (TG‒DSC) data were recorded within 30‒900 °C at a heating rate of 3 °C/min by STA (NETZSCH, STA449 F3 Jupiter®) with continuous air flow. Room temperature photoluminescence spectra of the powder samples were measured using a Hitachi F-7000 fluorescence spectrophotometer.
3 Results and discussion
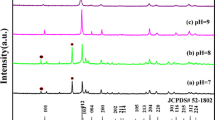
X-ray diffraction (XRD) was employed to identify the phase composition of the synthesized precursors. Figure 1 shows the XRD patterns of the precursors (S1‒S6) without heat treatment and the products (C1‒C6) after calcined. The results of Fig. 1a state that all the diffraction peaks of the samples synthesized at different conditions during solvothermal reaction are well indexed to the monoclinic yttrium oxide hydroxide nitrate Y4O(OH)9NO3 (JCPDS No. 79-1352). The similarity of XRD patterns of the different samples indicates that the phases of precursors are independent of the rare earth nitrates concentration and benzyl alcohol/1-octylamine volume ratio. However, the diffraction peaks of sample S5 are obviously broadening compared with those of samples S1, S2, S3, S4, and S6, suggesting that the degree of crystallization of sample S5 is lower than the other samples. This can be attributed to the fact that sample S5 consists of two dimensional nanosheets with reduced periodicity outside the plane direction [36]. As shown in Fig. 1b, all the diffraction peaks of the products (C1‒C6) after being calcined at 800 °C for 2 h are indexed to the cubic yttrium oxide Y2O3 (JCPDS No. 73-1334). The results indicate that precursors synthesized at different conditions have all converted to yttrium oxide with no residual impurities.

XRD patterns of samples, a S1‒S6, b C1‒C6
Figure 2 shows the EDS spectra of sample S1 and C1 (a), FT-IR spectra of sample S1, S4, S5 and C1, C4, C5 (b) and TG-DSC curves of sample S1 (c). The results of other samples are similar, and sample S1 and C1 are selected as examples to investigate the composition and avoid repetition. The atomic ratio of Re (Y, Eu) and O of sample S1 is 1:4.04, which is far from the theoretical value of Y4O(OH)9NO3:Eu3+ because of the organic residues adsorbing on the surface of the precursors. The atomic ratio of Re (Y, Eu) and O of sample C1 is 1:1.6 that is in great agreement with the theoretical value of Y2O3. The atomic ratio of Y and Eu is 96:4 which fitted well with the ratio of raw materials. The element Eu existing in the EDS spectrum and disappearing in XRD spectrum indicate an efficient substitute doping of Eu3+ ion in the Y2O3 structure.

EDS spectra of sample S1 and C1 (a), FT-IR spectra of sample S1-S3 and C1-C3 (b), TG-DSC curves of sample S1 (c)
The FT-IR spectra of sample S1, S4 and S5 display a strong absorption band at 3400‒3500 cm−1, which is attributed to the stretching vibration of O–H bond. The peaks at 687, 843, 1085 and 1371 cm−1 are assigned to the bending vibrations and the stretching vibrations of the NO3 − groups. These results can support the Y4O(OH)9NO3 structure of the precursor. Moreover, the peak at 1521 cm−1 corresponds to the asymmetrical vibration modes of carboxyl (–COO–) groups, which may originate from the absorption of CO2 from the atmosphere and organic residues from the solutions [30, 32]. The sharp absorption peak of samples C1, C4 and C5 at 564 nm results from the vibration of Y–O band. The weak absorption peaks at 3424 and 1635 cm−1 show the existence of water molecules on the surface of Y2O3:Eu3+.
As shown in Fig. 2c, the two major stages of weight loss with endothermic peaks at 384 and 516 °C are in great agreement with the decomposition of Y4O(OH)9NO3 into Y2O3 [30, 37]. However, the total weight loss of sample S1 during the measurement is 25.91% which deviates from the theoretical value (23.01%) calculated from the complete decomposition of Y4O(OH)9NO3 into Y2O3. The excess weight loss is due to the weight loss at a temperature below 300 °C, which can be explained by the absorption of organic residues and water.
Figure 3 shows the SEM and TEM images of samples C1, C5 and C6 with different rare earth concentrations. The size of the samples keeps increasing with the increment of the RE3+ concentrations (Fig. 3a, c, d). The TEM image indicates that the sample C5 consists of layered nanosheets whose size are about 30 nm (Fig. 3b), which shows no difference with the previous report [21]. Moreover, the nanosheets are overlapped and aggregated because of the large surface area and high surface energy. The sizes of the samples increase with the increment of rare earth concentrations, and the morphologies transform from nanosheets to sub-microspheres, and eventually to microrods. Figure 3c shows that the size of the sub-microspheres is about 500 nm. By further increasing the concentrations, the length and diameter of sample C6 are about 1.5 μm and 600 nm respectively. The results can be explained with the diffusion-limited aggregation (DLA) model. The size of nanosheets is small because of the low concentration which means that prime particles could difficultly cluster together for growth. The mean free path of the prime particles decreases resulting in faster growth rate and larger size when the concentrations increase. The strong driving force of random aggregation originate from the high surface energy of particles. The yttrium hydroxide nitrate [Y4O(OH)9NO3] tends to grow into one-dimensional morphology because of its intrinsical anisotropy.

SEM and TEM images of samples synthesized with different rare earth concentrations but the same benzyl alcohol/1-octylamine ratio, SEM and TEM images of sample C5 respectively (a, b), SEM images of sample C1 (c), and SEM images of sample C6 (d)
In general, the size and shape of the compound can be modulated by controlling the crystal nucleation and relative growth rate on particular crystal surfaces during crystal growth. To study the influence of the solvent, the samples synthesized at different volume ratio of benzyl alcohol/1-octylamine are shown in Fig. 4. The shapes of the samples evolve from sub-microspheres to microrods when decreasing benzyl alcohol volume. The yttrium hydroxide nitrate [Y4O(OH)9NO3] tends to grow into one-dimensional morphology because of its intrinsical anisotropy. However, the benzyl alcohol molecules could selectively adsorb onto specific crystal faces of Y4O(OH)9NO3 crystals to alter their surface energies, which could significantly decrease the growth rates of these surfaces and lead to isotropic growth. The sphere-like products appeared as final structures in Fig. 4a, which suggests that the benzyl alcohol volume is high enough to adsorb on the crystal faces in all directions to form isotropic growth. The length of the microrods increases with decreasing benzyl alcohol volume, which confirms this point of view.

SEM images of sample C1 (a), sample C2 (b), sample C3 (c) and sample C4 (d). The samples were synthesized at the same conditions except the benzyl alcohol/1-octylamine ratio
Figure 5 shows the photoluminescence excitation and emission spectra and decay curves of samples C1–C6 recorded at room temperature. By monitoring the characteristic emissions of Eu3+ ions at 613 nm, the excitation spectra of samples inserted in Fig. 5a show the broad band with the strongest peak at 245 nm which is due to the charge transfer band of from the 2p orbital of O2− to the 4f orbital of Eu3+. The emission spectrum shows that the peaks centered at 534, 581, 588, 593, 600, 613, and 631 nm are associated with the 5D1 → 7F1 and 5D0 → 7FJ (J = 0, 1, 1, 1, 2, 3) transition of Eu3+ ions, respectively. When adjusting the RE3+ concentrations, the samples with larger size emitting stronger red light indicate that the PL intensity may be associated with the size of the samples. The volume ratio of the solvent affects not only the morphologies but also the PL intensity. When increasing the volume of 1-octylamine, the PL intensities decrease obviously due to the hydroxyl quenching from 1-octylamine. This result illustrates that the hydrophilic 1-octylamine absorbed on the surface of the samples results in fluorescence quenching.

Presents the room-temperature photoluminescence emission and excitation spectra of the samples C1‒C6 obtained after calcinations
The fluorescent decay curves of the 5D0 → 7F2 transition of Eu3+ ions (corresponding to 613 nm) in Y2O3:Eu3+ are shown in Fig. 5b. All curves can be fitted to a single exponential function as I = A + I 0exp(− t/τ), where τ is the decay lifetime. By fitting, the lifetimes of samples C1‒C6 are 1.60, 1.61, 1.44, 1.49, 2.12 and 1.62 ms, respectively. Considering the instrumental erors, the lifetime is almost unchanged except for that of sample C5. Figure 6 shows the standard CIE coordinate-color graph and the intensity ratio of 5D0 → 7F2 to 5D0 → 7F1 for samples C1–C6. The CIE coordinates of samples have no obvious change when adjusting the synthesis conditions. To investgate the change of lifetime, the intensity ratios (R) of 5D0 → 7F2 to 5D0 → 7F1 transitions were calculated to detect the local environment of the Eu3+ ions. Eu3+ at C2 symmetry site contributes to the 5D0 → 7F2 transition, and Eu3+ at both C2 and C3i symmetry sites contribute to the 5D0 → 7F1 transition. The ratio of sample C5 is obviously larger than the other samples, meaning that the higher occupancy at C2 symmetry site. The result indicates that the local environment of the Eu3+ ions in Y2O3 nanosheets results in the increasing of decay lifetime.

Presents the standard CIE coordinate-color graph and the intensity ratio of 5D0 → 7F2 to 5D0 → 7F1 for samples C1–C6
4 Conclusions
In summary, shape-controlled and size-adjustable Y2O3:Eu3+ phosphors were successfully synthesized by a non-aqueous solvothermal method and subsequent calcine process without any assistance of the templates and surfactant. The composition and crystal structure of Y4O(OH)9NO3 precursors were confirmed by XRD, FT-IR and TG-DSC. The shape of samples transformed from sheet to sphere, and eventually to rod by just increasing the concentration of rare earth elements. The benzyl alcohol could absorb on certain crystal face inducing oriented growth. Moreover, the PL intensity of Y2O3:Eu3+ phosphors strongly depend on the sizes of the samples. Finally, this facile method acts as the link between morphologies and properties, which may be expected to bring new opportunities in the controllable synthesis of other inorganic functional materials.
References
S. Gai, C. Li, P. Yang, J. Lin, Recent progress in rare earth micro/nanocrystals: soft chemical synthesis, luminescent properties, and biomedical applications. Chem. Rev. 114, 2343–2389 (2014)
M. Wang, M. Li, A. Yu, J. Wu, C. Mao, Rare earth fluorescent nanomaterials for enhanced development of latent fingerprints. ACS Appl. Mater. Interfaces 7(51), 28110–28115 (2015)
H. Dong, S.-R. Du, X.-Y. Zheng, G.-M. Lyu, L.-D. Sun, L.-D. Li, P.-Z. Zhang, C. Zhang, C.-H. Yan, Lanthanide nanoparticles: from design toward bioimaging and therapy. Chem. Rev. 115(19) 10725–10815 (2015)
S. Takeshita, Y. Takebayashi, H. Nakamura, S. Yoda, Gas-responsive photoluminescence of YVO4:Eu3+ nanoparticles dispersed in an ultralight, three-dimensional nanofiber network. Chem. Mater. 28(23), 8466–8469 (2016)
X. Wang, P. Yan, Y. Li, G. An, X. Yao, G. Li, Highly efficient white-light emission and UV–Visible/NIR luminescence sensing of lanthanide metal–organic frameworks. Cryst. Growth Des. 17(4), 2178–2185 (2017)
J. Zhao, L. Wu, C. Zhang, B. Zeng, Y. Lv, Z. Li, Q. Jiang, Z. Guo, Highly efficient saturated visible up-conversion photoluminescent Y2O3: Er3+ microspheres pumped with a 1.55 μm laser diode. J. Mater. Chem. C 5(16), 3903–3907 (2017)
Y. Song, B. Shao, Y. Feng, W. Lü, J. Huo, S. Zhao, M. Liu, G. Liu, H. You, Emission enhancement and color tuning for GdVO4:Ln3+ (Ln = Dy, Eu) by surface modification at single wavelength excitation. Inorg. Chem. 56(1), 282–291 (2017)
H.Q. Wang, R.D. Tilley, T. Nann, Size and shape evolution of upconverting nanoparticles using microwave assisted synthesis. CrystEngComm 12(7), 1993–1996 (2010)
Q. Zhu, J.-G. Li, X. Li, X. Sun, Morphology-dependent crystallization and luminescence behavior of (Y,Eu)2O3 red phosphors. Acta Mater. 57(20), 5975–5985 (2009)
Y. Li, Y. Wang, Y. Song, X. Zhou, K. Zheng, Y. Sheng, H. Zou, Controlled synthesis and luminescence properties of GdF3 with different crystalline phases and morphologies. CrystEngComm 19(11), 1517–1527 (2017)
B. Liu, C. Li, P. Yang, Z. Hou, J. Lin, 808-nm-Light-excited lanthanide-doped nanoparticles: rational design, luminescence control and theranostic applications. Adv. Mater. 29, 1605434 (2017)
R. Meenambal, P. Nandha Kumar, P. Poojar, S. Geethanath, S. Kannan, Simultaneous substitutions of Gd3+ and Dy3+ in β-Ca3(PO4)2 as a potential multifunctional bio-probe. Mater. Des. 120, 336–344 (2017)
J. Xu, P. Yang, M. Sun, H. Bi, B. Liu, D. Yang, S. Gai, F. He, J. Lin, Highly emissive dye-sensitized upconversion nanostructure for dual-photosensitizer photodynamic therapy and bioimaging. ACS Nano https://doi.org/10.1021/acsnano.7b00944
S. Fukushima, T. Furukawa, H. Niioka, M. Ichimiya, T. Sannomiya, N. Tanaka, D. Onoshima, H. Yukawa, Y. Baba, M. Ashida, J. Miyake, T. Araki, M. Hashimoto, Correlative near-infrared light and cathodoluminescence microscopy using Y2O3:Ln, Yb (Ln = Tm, Er) nanophosphors for multiscale, multicolour bioimaging. Sci. Rep. 6, 25950 (2016)
G. Yuan, M. Li, M. Yu, C. Tian, G. Wang, H. Fu, In situ synthesis, enhanced luminescence and application in dye sensitized solar cells of Y2O3/Y2O2S:Eu3+ nanocomposites by reduction of Y2O3:Eu3+. Sci. Rep. 6, 37133 (2016)
J. Kaszewski, M.M. Godlewski, B.S. Witkowski, A. Słońska, E. Wolska-Kornio, Ł. Wachnicki, H. Przybylińska, B. Kozankiewicz, A. Szal, M.A. Domino, E. Mijowska, M. Godlewski, Y2O3:Eu nanocrystals as biomarkers prepared by a microwave hydrothermal method. Opt. Mater. 59, 157–164 (2016)
G. Chen, W. Qi, Y. Li, C. Yang, X. Zhao, Hydrothermal synthesis of Y2O3:Eu3+ nanorods and its growth mechanism and luminescence properties. J. Mater. Sci.: Mater. Electron. 27(6), 5628–5634 (2016)
J. Li, J.-G. Li, Q. Zhu, X. Sun, Room-temperature fluorination of layered rare-earth hydroxide nanosheets leading to fluoride nanocrystals and elucidation of down-/up-conversion photoluminescence. Mater. Des. 112, 207–216 (2016)
S. Tao, Y. Li, G. Chen, X. Zhao, Critical temperature of smart meta-superconducting MgB2. J. Supercond. Nov. Magn. 30(6), 1405–1411 (2017)
Z. Zhang, S. Tao, G. Chen, X. Zhao, Improving the critical temperature of MgB2 superconducting metamaterials induced by electroluminescence. J. Supercond. Nov. Magn. 29, 1159–1162 (2016)
W. Cheng, F. Rechberger, M. Niederberger, Three-dimensional assembly of yttrium oxide nanosheets into luminescent aerogel monoliths with outstanding adsorption properties. ACS Nano 10, 2467–2475 (2016)
G. Chen, S. Tao, C. Yang, X. Zhao, Facile preparation and fluorescence enhancement of mesoporous Eu-doped-Y2O3 phosphors. J. Mater. Sci.: Mater. Electron. 26, 5970–5974 (2015)
L. Wang, H. Fang, H. Xu, C. Wang, Y. Li, Y. Liu, S. Zhong, Ionic liquid-assisted preparation of square-shaped Y2O3 nanoplates. Mater. Res. Bull. 61, 89–94 (2015)
C. Cascales, C. Zaldo, F. Esteban-Betegón, R. Calderón-Villajos, From porous to dense Tm3+–Lu2O3 micro- and nanosized crystalline morphologies designed through hydrothermal precursors: assessment on infrared emission properties. CrystEngComm 14(10), 3577 (2012)
B. Zhao, X. Xie, S. Xu, Y. Pan, B. Yang, S. Guo, T. Wei, H. Su, H. Wang, X. Chen, V.P. Dravid, L. Huang, W. Huang, From ScOOH to Sc2O3: phase control, luminescent properties, and applications. Adv. Mater. 28(31), 6665–6671 (2016)
Y. Wang, T. Wei, X. Cheng, H. Ma, Y. Pan, J. Xie, H. Su, X. Xie, L. Huang, W. Huang, Insights into Li+-induced morphology evolution and upconversion luminescence enhancement of KSc2F7:Yb/Er nanocrystals. J. Mater. Chem. C 5(14), 3503–3508 (2017)
S. Zeng, K. Tang, T. Li, Z. Liang, 3D flower-like Y2O3:Eu3+ nanostructures: template-free synthesis and its luminescence properties. J. Colloid Interface Sci. 316(2), 921–929 (2007)
Y. Zheng, H. You, G. Jia, K. Liu, Y. Song, M. Yang, Y. Huang, H. Zhang, Facile synthesis of Y4O(OH)9NO3:Eu3+/Y2O3:Eu3+ nanotubesand nanobundles from nanolamellar precursors. CrystEngComm 12, 585–590 (2010)
N. Zhang, X. Liu, R. Yi, R. Shi, G. Gao, G. Qiu, Selective and controlled synthesis of single-crystalline yttrium hydroxide/oxide nanosheets and nanotubes. J. Phys. Chem. C 112, 17788–17795 (2008)
Q. Zhao, H. You, W. Lü, N. Guo, Y. Jia, W. Lv, B. Shao, M. Jiao, Dendritic Y4O(OH)9NO3:Eu3+/Y2O3:Eu3+ hierarchical structures: controlled synthesis, growth mechanism, and luminescence properties. CrystEngComm 15(24), 4844–4851 (2013)
J. Dai, M. Lv, G. Li, X. Li, Synthesis and luminescence properties of highly uniform SiO2@LaPO4:Eu3+ core–shell phosphors. Mater. Des. 83, 795–800 (2015)
G. Jia, H. You, L. Zhang, Y. Zheng, K. Liu, Y. Huang, H. Zhang, Facile synthesis of highly uniform octahedral LuVO4 microcrystals by a facile chemical conversion method. CrystEngComm 11(12), 2745–2750 (2009)
E. Pavitra, J. Su Yu, A facile large-scale synthesis and luminescence properties of Gd2O3:Eu3+ nanoflowers. Mater. Lett. 90, 134–137 (2013)
Y. Song, H. Zou, Y. Sheng, K. Zheng, H. You, 3D hierarchical architectures of sodium lanthanide sulfates: hydrothermal synthesis, formation mechanisms, and luminescence properties. J. Phys. Chem. C 115(40), 19463–19469 (2011)
S.K. Hussain, G. Nagaraju, E. Pavitra, G. Seeta Rama Raju, J.S. Yu, La(OH)3:Eu3+ and La2O3:Eu3+ nanorod bundles: growth mechanism and luminescence properties. CrystEngComm 17(48), 9431–9442 (2015)
N. Pinna, G. Garnweitner, P. Beato, M. Niederberger, M. Antonietti, Synthesis of yttria-based crystalline and lamellar nanostructures and their formation mechanism. Small 1, 112–121 (2005)
E. Yang, G. Li, C. Fu, J. Zheng, X. Huang, W. Xu, L. Li, Eu3+-doped Y2O3 hexagonal prisms: shape-controlled synthesis and tailored luminescence properties. J. Alloys Compd. 647, 648–659 (2015)
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51272215, 11674267) and the National Key Scientific Program of China (under Project No. 2012CB921503).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, G., Li, Y., Qi, W. et al. Morphology-tailored synthesis and luminescent properties of Y2O3:Eu3+ phosphors. J Mater Sci: Mater Electron 29, 2841–2847 (2018). https://doi.org/10.1007/s10854-017-8213-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-8213-7