
Abstract
This paper highlights on the consequence of replacing tetravalent Sn4+ ions of the SnO2 by divalent Mn2+ ions on their structural, optical and magnetic properties. Samples of Sn1−xMnxO2 with x = 0.01, 0.02, 0.03 and 0.04 were synthesized using microwave irradiated solvothermal process. The X-ray powder diffraction patterns reveal the rutile tetragonal phase of all doped SnO2 samples with no secondary phases. The transmission electron microscopy results show the formation of spherical nanoparticles of size 10–16 nm. Morphological changes were observed by scanning electron microscopy. The functional groups were investigated using Fourier Transform Infrared Spectroscopy studies. Optical studies were carried by UV–Vis Spectroscopy and Fluorescence Spectroscopy. Electron Paramagnetic resonance was used to calculate the Lande splitting factor ‘g’. The magnetic properties were using Vibrating Sample Magnetometer. SnO2 with lower Mn doping shows ferromagnetism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nanotechnology is the most essential and electrifying forefront field in Physics, Chemistry, Engineering and Biology. As unique optical, chemical, and magnetic properties of low dimensional inorganic semiconducting metal oxide nanomaterials were deeply influenced by the size and morphology of the nanostructures. The preparation of nano sized crystallites with different sizes provided an opportunity to explore the possible changes in their physical and chemical properties with size and shape [1,2,3].
Tin oxide (SnO2) is one of the n-type wide band-gap semiconductor material (3.6 eV) and has large exciton binding energy (130 meV) [4]. SnO2 evinces interest because it is a naturally non-stoichiometric prototypical transparent conducting oxide. It has a high band gap of almost 4 eV, plasma frequency in the IR region and, when suitably doped, can be used both as a p-type and n-type semiconductor. It crystallizes in the tetragonal rutile type of structure, D144h (P42/mnm) with two Sn and four oxygens per unit cell. The lattice parameters are a = b = 0.4737 nm, c = 0.3185 nm and c/a = 0.673 [5].
As an n-type semiconductor, SnO2 shows very high sensitivity towards H2, CO, hydrocarbon, and alcohol. It combines the low electrical resistance region. This property makes it a noticeable applicant for optoelectronic applications. The optoelectronic properties such as photoluminescence and optical band gap of SnO2 can also be improved by impurity doping. It has been used as a solid state sensor mainly due to its sensitivity towards different gaseous species [6, 7], photovoltaic energy conversion [8], to make transparent conductive thin film coatings [9], solar cells [10] etc. SnO2 nanoparticles drag scrutiny due to its promising applications in gas sensors [11], microelectronics [12], solar cells [13] and photoelectrochemistry [14], lithium cells [15] and photocatalysts [16]. The high chemical stability, low cost, nontoxicity and excellent electro-optical properties make it suitable for numerous technological applications [17]. Another property of transparent conducting oxides (TCOs) such as SnO2 is that they are transparent in the visible region and are highly reflective in the infrared region [18]. This property is responsible for today’s dominant use of SnO2 as an energy conserving material [18]. SnO2 coated architectural windows, for instance, allow transmitting light, but keeping the heat out in the building depending on the climatic region [18].
Magnetic properties of tin oxide nanoparticles are used in magnetic data storage and magnetic resonance imaging. Nanoparticles discover widespread uses in several fields such as microelectronics, photo catalysis, nonlinear optics and photoelectro chemistry. Recently transition metal doped SnO2 has gained much attention due to the possibility of exhibiting room-temperature ferromagnetism. Diluted magnetic semiconductor (DMS) utilizes, synchronously, carrier charge and spin degrees of freedom [19]. Their interaction is expected to exhibit a novel physical character to be applied in new devices. The control of spin dependent phenomena in electronic oxides or more conventional semiconductors may lead to devices such as spin light-emitting diodes (spin-LEDs), spin field effect transistors (spin-FETs) and so on [19]. DMS is expected to play an important role in materials science and future spintronics.
Kimura et al. obtained a rutile type oxide diluted magnetic semiconductor with high Mn concentrations (x < 0.34) with room temperature paramagnetic behaviour [20]. Due to its larger thermal solubility (10 mol%) Mn can increase the amount of injected spins and carriers make Mn doped SnO2 as a promising material for spintronics applications [21, 22]. Brahma et al. [23] verified good transparency and conductivity in Mn-doped SnO2 films deposited by thermal evaporation. Fitzgerald et al. [24] shows ferromagnetism in terms of carrier mediated exchange in spin split impurities, Manganese is an efficient luminescent activator in a number of host lattice, e.g., silicates, sulfides, and fluorides of zinc or cadmium [25]. The intra-d-shell transition in Mn2+ is sensitive to the magnitude of the crystal field and the luminescent wavelength can be broadly tuned from green to orange or red according to the coordination environment [26]. Gopinadhan et al. reports room temperature ferromagnetism in manganese doped SnO2 thin films prepared by spray pyrolysis method [27]. Tian et al. synthesized Mn doped tin dioxide nanoparticles by chemical co-precipitation method and report that manganese ions incorporate in the tin dioxide lattice as a mixture of Mn3+ and Mn4+. Room temperature ferromagnetism is observed for samples with Mn concentrations under 5 at.% and calcined at 450 °C with a strong correlation to the surface structural defects [28]. Azam et al. investigated the electrical properties of Mn doped tin oxide nanoparticles obtained via the sol–gel method [29]. Manganese doped tin oxide thin films obtained through the spray pyrolysis method were studied by Vadivel et al. [30]. Sabri et al. shows the effect of Mn doping on structural and optical properties of SnO2 nanoparticles prepared by mechanochemical processing [31]. Saravanakumar et al. [32] prepared shows the effect of Mn doping on the structural, optical and magnetic properties of SnO2 nanoparticles via co-precipitation method. SnO2 with lower Mn content shows larger magnetization. The Mn doped SnO2 nanoparticles synthesized by chemical co-precipitation method has been investigated by Agrahari et al. [33]. All the doped SnO2 nanoparticles show paramagnetic behavior at room temperature. Salah et al. [34] shows the structural, optical and electrical properties of Mn-doped SnO2 nanoparticles via the microwave technique.
Microwave dielectric heating not only improves the rate of formation, it also increases the material quality, size distributions, accelerating the reaction rate, yield improvement, quicker reaction time, smaller particle size, high purity materials, and enhanced physicochemical properties [35]. This method allows volumetric, selective and uniform heating which improves the physical and mechanical properties [36].
Various performances of SnO2 are due to the presence of native defects such as oxygen vacancies, tin vacancies, tin interstitials and oxygen interstitials. The nature of the defects strongly depends on the synthesis method and the dimension of the material. Based on the preparation state, the types and numbers of defects, morphology and light emitting properties may vary. The ferromagnetism in SnO2 nanostructures has been attracting wide attention in current years, since it can associate the properties of spintronics with nanostructures The origin of FM in Mn doped SnO2 materials has been highly debated during the recent past. In this work we report the room temperature ferromagnetism via a solvothermal method for the Mn doping concentrations of 1% and 2, 3, and 4% show paramagnetism. We have performed the comprehensive experimental studies of the Mn-doped SnO2 powders which have provided a strong support for “intrinsic” origin of ferromagnetism in the 1% Mn-doped SnO2 powders, that is ascribed to the presence of oxygen vacancy defects. The nature of the structural defects present in doped samples was identified by FL and EPR studies.
2 Materials and methods
Analytical grade precursor materials such as Tin(II) chloride dihydrate (SnCl2·2H2O) (Merck), Manganese(II) acetate, urea [H2NCONH2] were used to synthesize manganese doped SnO2 nanoparticles by solvothermal method. To synthesize Sn1−xMnxO2 (x = 0.01, 0.02, 0.03, and 0.04) samples, the stoichiometric amount of Tin(II) chloride dehydrate, Manganese(II) acetate and urea was added to ethylene glycol [CH2OH·CH2OH]. The ensuing solution was mixed thoroughly. The solution so attained was stirred to homogeneity at room temperature using a magnetic stirrer for 1 h and kept in a microwave oven. A domestic microwave oven (M/O S20M WW-CG) operating at 2.45 GHz was used as the device. The microwave oven was operated at 1 min/cycle and cooled in between the cycles. Microwave heating was carried through until the solvent become dry. The content was allowed to cool and transferred to a beaker. It was washed thrice with double distilled water to remove inorganic impurities. Then the settled down powder was filtered using Whatmann filter paper and the collected powder has allowed to dry. Again the product was washed using acetone to remove unwanted organic impurities and dried for 2 h. The synthesized nanoparticles were filtered, dried in 5 sunny days and annealed in a muffle furnace at 500 °C for an hour. The final product was taken out in the nice powder form. Figure 1 shows the procedure for the synthesis of Mn doped SnO2 nanoparticles. Table 1 shows the concentrations of precursors used.

Synthesis of Mn doped SnO2 nanoparticles
The X-ray diffraction (XRD) patterns of the nanopowder were taken using an X-ray diffractometer (PANalytical X’Pert PRO) with Cu radiation (λ = 0.15418 nm) in the region of 10°–80° with step size 0.01°. The morphology of the sample was observed by scanning electron microscopy (SEM, Carl Zeiss EVO 18 model). TEM images were taken in PHILIPS CM200. The occurrence of functional groups in Mn doped SnO2 nanoparticles was studied by Fourier Transform Infra Red (FTIR) spectrometer (Model: Perkin Elmer, Make: Spectrum RX1) in the range of 4000–400 cm−1. Optical measurements were carried out using UV–Vis spectrophotometer and luminescence spectrophotometer (Model: Lambda 35, Make: Perkin Elmer). Electron Spin Resonance Spectrometer (JES FA200) used for the analysis of species that contain unpaired electrons. Room temperature magnetic measurement was carried out using a vibrating sample magnetometer VSM (Lakeshore- 7410). All measurements were performed at room temperature.
3 Results and discussion
3.1 Structural properties
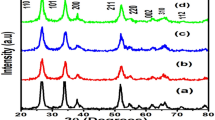
Figure 2a shows the XRD patterns of Sn1−xMnxO2 nanoparticles annealed at 500 °C. The XRD pattern shows that Mn doping did not change the tetragonal rutile structure of SnO2 nanoparticles. The diffraction peaks located at 2θ are 26.59°, 33.85°, 37.95°,51.78°, 54.78°, 57.79°, 61.92°, 64.7°, 65.94°, 71.31° and 78.72° for the lattice planes (110), (101), (200), (211), (220), (002), (310), (112), (301), (202) and (321). The patterns are in good agreement with SnO2 tetragonal rutile structure of JCPDS file #770447. Peaks due to tin metal and Mn were not detected, thus showing high purity of the product. A careful observation of the enlarged view of (211) peak for all the doped samples, as presented through Fig. 2b indicates the shift in peak positions. Shifting for every peak may due to lattice contraction or lattice expansion depend on how we synthesize the nanoparticles. Shifting could come from at surface with less D-spacing due to surface strain or systematic error from the detector. The diffraction peaks are observed to have the slight shift of the peak position towards higher angles with increased Mn content imply that the Mn atoms effectively occupy the Sn site without changing the rutile structure.

a XRD spectra of the Sn1−xMnxO2 nanoparticles, b shift of the (211) XRD peaks of the Sn1−xMnxO2 nanoparticles
Crystalline sizes in different directions are estimated in terms of Debye–Scherrer equation [37]
where D is the crystalline size, K is a constant (0.9 assuming that the particles are spherical), λ is the wavelength of the incident X-rays (0.1546 nm), β is the full width at half maximum (FWHM), θ is the Bragg diffraction angle. The lattice constants (a = b, and c) are estimated by [37]
where d is the lattice spacing and h, k and l are Miller indices. Tables 2, 3, 4 and 5 shows the lattice parameters, interplanar distance, Bragg diffraction angle, full width at half maximum and size of Sn0.99Mn0.01O2, Sn0.98Mn0.02O2, Sn0.97Mn0.03O2 and Sn0.96Mn0.04O2 nanoparticles respectively. Table 6 shows the crystalline sizes and the lattice parameters of the Sn1−xMnxO2 nanoparticles at 500 °C.
The lattice parameters a, c and unit cell volume exhibited decreasing when the Mn doping level has increased to 4 at.%. However, the cell volume of the tetragonal phase of Mn doped samples was found to decrease from 72.115 to 71.032 Å3 may be due to the decrease of defects. Particle size is minimum at doping level of 2%. The value of c/a parameter 0.67 is nearby to the standard value of tetragonal structure. The crystallite sizes of Al doped SnO2 nanoparticles are found to be in the range of 10–16 nm. All these variations in a, c, V and size indicate that the doping of Mn took place in SnO2 lattice. Figure 3 shows the lattice parameters a, c, cell volume V and average crystallite size of particles as a function of doping concentration. Figure 3 shows the lattice parameters a, c of particles as a function of doping concentration.

The lattice parameters a, c, cell volume V and average crystallite size of Mn doped SnO2 particles as a function of doping concentration
The SEM images of (a) Sn0.99Mn0.01O2 and (b) Sn0.96Mn0.04O2 samples in Fig. 4 shows the morphology and size distribution of particles. SEM pictures have been magnified to the two same magnification, which is useful when matching the sizes of the crystallites of the samples. The size of the Sn0.99Mn0.01O2 was estimated to be around 14.47 nm using XRD. Also the size of the particle was valued to be around 14.21 nm using XRD. However, a small aggregation of particles has been perceived in the Sn0.96Mn0.04O2 sample as related to Sn0.99Mn0.01O2 sample. However, on the whole, it was clear that doping can inhibit the excess aggregation of SnO2 nanoparticles. This specifies that the diffusion of Mn2+ into the SnO2 lattice can change the surface character of the primary nanoparticles. Figure 5 displays the EDAX spectrum of Sn0.96Mn0.04O2 samples. The EDAX spectrum of the sample clearly displays the existence of Sn, Mn and O elements, revealing that the manganese atoms were indeed doped in the SnO2 matrix.

SEM images of Sn0.99Mn0.01O2 and Sn0.96Mn0.04O2 samples

EDAX image of Sn0.96Mn0.04O2 sample
Morphology of Mn doped SnO2 nanoparticles was investigated by FETEM. Figure 6 shows the TEM of Sn0.96Mn0.04O2 sample annealed at 500 °C. It is observed that these nanoparticles are nearly tetragonal in shape with uniform particle size of diameter ranging from 8 to 16 nm and this outcome is in agreement with the particle size of 16 nm obtained from the XRD pattern.

TEM image of Sn0.96Mn0.04SnO2 sample
Figure 7 shows the Selected Area Electron Diffraction patterns (SAED) of Sn0.96Mn0.04O2 sample annealed at 500 °C. SAED pattern shows continuous ring patterns without any additional diffraction spots and rings of secondary phases which enlighten the good crystalline nature. The distance of each ring from the centre and its reciprocal values gives the interplanar distance ‘d’. XRD results are confirmed by the combined study of these SEM and TEM studies.

SAED pattern of Sn0.96Mn0.04SnO2 sample
3.2 Fourier transformed infrared spectroscopy (FTIR)
It is used to study the absorption properties of doped samples and hence to conclude the nature of bonds present in the doped samples.
Figure 8 shows the FTIR spectra of Mn doped SnO2 nanoparticles annealed at 500 °C in the wave number range of 4000–400 cm−1. The doped samples have similar profiles. The intense and broadband from wave number 3400–3900 and 3444 cm−1 can be attributed to the O–H vibration of absorbed water on the sample surface [37]. The samples show a band around 650 cm−1. It is mainly ascribed to the strong metal oxygen vibration (Sn–O–Sn) frequency [38], which authorizes the formation of the SnO2 nanoparticles. The band around 482 cm−1 is ascribed to symmetric Sn–O–Sn frequency and Mn–O bonds [39]. Table 7 shows the functional groups present in aluminium doped SnO2 nanoparticles.

FTIR spectra of Sn1−xMnxO2 nanoparticles
3.3 Optical studies
3.3.1 UV–Vis spectroscopy
The direct band gaps of the nanoparticles were determined from the Tauc relation [40] given by Eq. (3). For semiconductor nanoparticles, following equation has been used to relate absorption coefficient with incident photon energy
where Eg is the band gap energy, hν is the incident photon energy, K is a constant, α(ν) is absorption coefficient which can be defined by the Beer–Lambert’s law as follows:
where A is the absorbance, ρ is the density of the SnO2 nanoparticles, c is the concentration and l is the path length. The exponent ‘n’ in Eq. (3) depends on the type of the transition and n may have values 1/2, 2, 3/2, 3 for allowed direct, allowed indirect, forbidden direct and forbidden indirect transitions respectively. In case of SnO2 nanoparticles the value of n is 1/2 for allowed direct transition. Therefore, by plotting (αhν)2 versus hν and by extrapolating the curve to zero absorption coefficient band gap (Eg) can be determined using Eq. (3). Figure 9 shows the tauc plot of (αhν)2 versus hν. When (αhν)2 is zero, the photon energy is Eg. The band gap values of the Mn doped samples were found to be 5.07, 4.5, 2.68 and 2.72 eV for the Sn1−xMnxO2 samples x = 0.01, 0.02, 0.03 and 0.04 respectively. The band gap energy of Mn-doped SnO2 is seen to decrease with the Mn doping, achieves a minimum value of 2.68 eV at x = 0.03, and with further addition of doping (x = 0.04) energy gap increased. It has been witnessed that the band gap reduces for low concentration doping and increases for higher concentration as expected on the basis of virtual crystal approximation (VCA) because of the band gap of the MnO ~4.2 eV. In many DMS systems such deviation from the linear monotonic increase in the form of “band gap bowing”. For the low concentration Mn doping the reduction in the band gap may be due to exchange interaction between d electrons of the Mn ions and the s and p electron of the host band. It is noted that the energy band gap value is decreasing with increasing Mn content, except for the sample Sn0.96Mn0.04O2. It may due to the creation of holes because of lower valency of manganese. The decreasing in the band gap was attributing to the presence of holes. Each substitution Mg atom will create two holes in the O 2p state because of the lower-valence than Sn+4. They form the impurity band inside the forbidden gap region. The electrons in the valance band are excited, and they can be combined with the holes in the impurity band first. However, they are not stable. The trapped electrons will recombine with the hole in the conduction band, resulting the shrinking of the band gap [41]. The obtained band gap of 4 mol% Mn doped SnO2 nanoparticles was found to be 2.72 eV. This increase might be due to the formation of additional electronic levels inside the band gap of SnO2 by Mn4+ ions. These electron levels were expected to cause charge transfer transitions between the conduction/valance band of SnO2 and Mn ions [34].

Band gap values of Sn1−xMnxO2 nanoparticles
3.3.2 Fluorescence spectroscopy
Defects such as tin interstitials, dangling bonds and oxygen ion vacancies introduce the energy states in the band gap act as an origin of photoluminescence [42]. In luminescence studies, high photon excitation energies above the band gap can be the most effective for bulk materials. But the efficiency of the luminescence decreases at high excitation energy for nanoparticles. Each particle size emits light at a characteristic frequency so the fluorescence emission spectrum reflects the emission from the overall particle size distribution. Due to their very small nanosize, the nanoparticles have susceptibility to various surface defects. The fluorescence spectra exposed the defect related emission. Figure 10 shows the emission spectra of Sn1−xMnxO2 nanoparticles using 270 nm as the excitation wavelength.

Fluorescence emission spectra of Sn1−xMnxO2 nanoparticles
Figure 10 presents the room temperature spectra fluorescence emission of Sn1−xMnxO2 NPs recorded in the range of 300–700 nm. It can be seen that the spectra are sharp and symmetric with a center at approximately 547 nm. There is an emission peak centred in all the samples at wavelength 547 nm, in the green region, with the corresponding energy of 2.26 eV. In the UV region, PL spectra of SnO2 exhibited an emission band at 372 nm (3.33 eV). Since this emission energy is much lower than the energy band gap of SnO2, the visible emission cannot be assigned to the direct recombination of conduction electron to the holes in valence band; instead, they are assigned to the defect-related emission. It is reported that the SnO2 nanostructures can exhibit emission bands between 400 and 600 nm, because of the presence of different crystalline defects [43,44,45]. The sharp intensity of the green emission suggesting that more number of Mn ions is in tetrahedral sites. The emission process is credited to d-level spin-forbidden transition for the Mn2+ ions acting as an activating center. In particular, the transition from the lowest excited state to the ground state, is directly responsible for the green light emission. Sharp peaks of the PL spectra of samples probably due to limited energy states positioned within the band gap. The defects and impurities present in the sample contribute to emission in the visible region. In SnO2, the oxygen vacancies are present in three different charge states: Vo 0, Vo +, and Vo ++, in which Vo 0 is a very shallow donor [46]. Among the different types of oxygen vacancies, the 547 nm peak related to single oxygen vacancies (Vo+) [47, 48]. In fact, VO + can easily trap a photoexcited electron through a non-recombination step to form the neutral VO x center, which is unstable at room temperature.
3.3.3 Electron paramagnetic resonance (EPR) spectroscopy
Electron paramagnetic resonance spectroscopy is an accessible dopant specific spectroscopic technique that probes transitions within Zeeman-split ground states of paramagnetic ions. For the usual transverse experimental configuration, the microwave absorption selection rule is MJ = ± 1. Because of zero-field splittings, transverse EPR is sensitive almost exclusively to ions with Kramers ground states, such as Mn2+ [49]. The EPR spectroscopy is an effective technique to investigate many types of transition metal ions, free radicals, biradicals, and different kinds of structural defects, particularly for paramagnetic materials. When the unpaired electron is placed within the applied magnetic field H0 two possible spin states of the electron will have different energies by the result of the Zeeman effect. The lower energy state occurs when the magnetic moment of the electron is aligned with the magnetic field and a higher energy state occurs when magnetic moment is aligned against the magnetic field. If a second weak alternating magnetic field H1 oscillating at a microwave frequency is applied right angles to the static field H0, then the electron will begin to precess in the axis of H1 and when the microwave frequency will equal to the precession frequency the resonance condition will be satisfied [50]. The following equation is used to calculate the g factor [51]:
where H is the static field (Gauss), γ is the microwave radiation frequency (Hz), B is the Bohr magnetron equal to 9.274 × 10−21 erg/Gauss, and h is Planck’s constant, 6.626 × 10−27 ergs/cycle.
Figure 11 shows the EPR spectrum of Mn2+ ions in Sn1−xMnxO2 nano powder recorded at room temperature. The rutile crystal structure of semiconducting SnO2 hosts a variety of defects, such as oxygen vacancies and Sn interstitials [52]. In Sn0.99Mn0.01O2, a broad signal without any six-line structure with g factor 1.9908 is typical for exchange-coupled Mn ions [49]. It is possible that the paramagnetic centers detected by EPR signal can be the origin of visible emission [53]. Ozcan et al. [54] have shown that the Sn4+, Sn2+, Vo 0 and Vo 2+ defect states have an even number of electrons and these are ESR silent singlets. The g factor around 2.0 can appear because of the Vo + defects and this defect is only ESR active [54].

EPR spectra of Sn1−xMnxO2 nanoparticles
In Sn0.96Mn0.04O2 sample Mn2+ ion, is unique among dn configurations and there is only one state with maximum spin multiplicity (6S5/2) which splits into three Kramers doublets (±5/2, ±3/2 and ±1/2). In the presence of magnetic field, the degeneracy is completely removed and five fine structure transitions are possible. Each fine structure transition will be split into six hyperfine components due to 55Mn hyperfine coupling and may give in all 30 allowed transitions. The sextets belong to the transitions 5/2 ↔ 3/2, 3/2 ↔ 1/2, 1/2 ↔ −1/2, −1/2 ↔ −3/2, −3/2 ↔ −5/2 [55]. The sextet observed at the central transition corresponding to 1/2 ↔ −1/2 transition has a g value of g ≈ 1.9971. Although the characteristic six-line hyperfine spectrum of Mn2+ was observed in the Sn0.96Mn0.04O2 sample, Sn0.98Mn0.02O2 and Sn0.97Mn0.03O2 show that this EPR signal disappeared entirely.
3.4 Magnetic property
3.4.1 Vibrating sample magnetometer (VSM)
We have done VSM analyses to observe magnetic characteristics of our fabricated samples. The origin of FM in Mn doped SnO2 materials has been highly debated during the recent past. Figure 12 shows M–H hysteresis loops of Mn doped SnO2 nanoparticles. The appearance of ferromagnetism in Sn1−xMnxO2 shows the clarification of ferromagnetic origin in DMS materials. The coercivity, retentivity, magnetization and particle size of Mn doped SnO2 nanoparticle are given in the Table 8.

M–H hysteresis loops of Mn doped SnO2 nanoparticles
The Sn0.99Mn0.01O2 sample shows larger saturation magnetization and with increasing Mn content, the ferromagnetization decreases. No impurity peaks were spotted in XRD pattern, any impurity or secondary phase is not responsible for observed ferromagnetism in Mn doped SnO2 samples. In the Sn0.99Mn0.01O2 sample most of the Mn2+ ions occupy substitutional position. Mn2+ ions, on substitution, create oxygen vacancies. Doping of Mn increases the number of oxygen vacancies and therefore an exchange interaction of the Mn ions via oxygen vacancies may induce ferromagnetism [32]. It shows the comprehensive experimental studies of the Mn-doped SnO2 powders which have provided a strong support for “intrinsic” origin of ferromagnetism in the 1% Mn-doped SnO2 powder is due to the presence of oxygen vacancy defects. With the help of EPR result, we have shown that Vo + is the main element that is responsible for RTFM in Sn0.99Mn0.01O2 sample. The sample shows high magnetic moment value due to the high oxygen vacancy concentration. The occurrence of the very high oxygen vacancy concentration gives rise to the ferromagnetic ordering. Although oxygen vacancies can be expected to be more at 2, 3 and 4%, the interaction of Mn2+ via oxygen vacancies is less pronounced. Therefore, both oxygen vacancies and an optimum level of Mn (with entire Mn2+ taking part in BMP) are necessary for obtaining ferromagnetism. High concentration of Mn contributes only to paramagnetism [32].
4 Conclusion
In conclusion, the effective synthesis of high purity Mn transition metal ions doped tin oxide (Sn1−xMnxO2, 0.01 ≤ x ≤ 0.05) nanoparticles has been achieved via the solvothermal method of microwave irradiation technique. The crystalline size was calculated from the Debye–Scherer formula, which proposed the formation of rutile tetragonal phase of all doped SnO2 nanoparticles, and this was further confirmed by the TEM and SAED patterns. The morphologies of the samples were detected from the SEM images, which display that most of the particles were near about spherical in shape. The EDX spectra show that the Mn ion is successfully incorporated into the SnO2 nanoparticles. The functional groups of the samples have been identified from FTIR. Optical studies have been carried out, using optical absorbance and fluorescence spectroscopies. The fluorescence spectra show a sharp and strong intensity of green emission peaks were observed in all the doped samples. EPR signal for the Sn0.99Mn0.01O2 nanoparticles suggesting the presence of singly ionized oxygen vacancies. EPR spectra of Sn0.96Mn0.04O2 exhibit hyperfine structure due to interaction of electron spin (S = 5/2) with its nuclear spin (I = 5/2) and the each fine structure transition which will be split into six hyperfine components due to 55Mn hyperfine coupling. VSM of shows Sn0.99Mn0.01O2 sample shows ferromagnetism due to exchange interactions between localized electron spin moments resulting from oxygen vacancies at the surfaces of nanoparticles. High concentration of Mn contributes only to paramagnetism.
References
S. Das, S. Kar, S. Chaudhuri, J. Appl. Phys. 99, 114303 (2006)
M.S. Gudiksen, L.J. Lauhon, J. Wang, D.C. Smith, C.M. Lieber, Nature 415, 617–620 (2002)
W. Gopel, K.D. Schierbaum, Sens. Actuators B 1, 26–27 (1995)
B. Liu, C.W. Cheng, R. Chen, Z.X. Shen, H.J. Fan, H.D. Sun, J. Phys. Chem. C 114, 3407–3410 (2010)
R. Brahma, M. Ghanashyam Krishna, A.K. Bhatnagar, Bull. Mater. Sci. 29(3), 317–322 (2006)
N.G. Deshpande, Y.G. Gudage, R. Sharma, J.C. Vyas, J.B. Kim, Y.P. Lee, Sens. Actuators B 138, 76–84 (2009)
V. Krivetskiy, A. Ponzoni, E. Comini, S. Badalyan, M. Rumyantseva, A. Gaskov, Electroanalysis 22, 2809–2816 (2010)
H.J. Snaith, C. Ducati, Nano Lett. 10, 1259–1265 (2010)
J.C. Manifacier, Thin Solid Films 90, 297–308 (1982)
GhassanE Jabbour, David Doderer, Nat. Photonics 4, 604–605 (2010)
R.S. Niranjan, Y.K.H. Wang, D.K. Kim, S.H. Jhung, J.S. Chang, I.S. Mulla, Mater. Chem. Phys. 92, 384 (2005)
S. Schiller, U. Heisig, K. Goedicke, H. Bilz, K. Steinfelder, Thin Solid Films 92, 81 (1982)
S.C. Lee, J.H. Lee, T.S. Oh, Y.H. Kim, Sol. Energy Mater. Sol. Cells 75, 481 (2003)
T Stergiopoulos, I.M. Arabatzis, H. Cachet, P. Falaras, J. Photochem. Photobiol. A 155, 163 (2003)
D. Aurbach, A. Nimberger, B. Markovasky, E. Levi, E. Sominsky, A. Gedanken, Chem. Mater. 14, 4155 (2002)
M. Miyauchi, A. Nikajima, T. Watanabe, K. Hasimoto, Chem. Mater. 14, 2812 (2002)
S. Gupta, B.C. Yadav, P.K. Dwivedi, B. Das, Condens. Matter Mater. Sci. arXiv:1208.4094 (2012)
O. Wurzinger, G. Reinhardt, Sens. Actuators B 103, 104–110 (2004)
S.J. Pearton, W.H. Heo, M. Ivill, D.P. Norton, T. Steiner, Semicond. Sci. Technol. 19, 59 (2004)
H. Kimura, T. Fukumura, M. Kawasaki, K. Inaba, T. Hasegawa, Appl. Phys. Lett. 80(1), 94 (2002)
P. Sharma, A. Gupta, F.J. Owens, A. Inoue, K.V. Rao, J. Magn. Magn. Mater. 282 115–121 (2004)
Y.S. Wang, P.J. Thomas, P. O’Brien J. Phys. Chem. B 110 21412–21415 (2006)
R. Brahma, M Ghanashyam Krishna, A.K. Bhatnagar, Bull. Mater. Sci. 29(3), 317 (2006)
C.B. Fitzgerald, M. Venkatesan, L.S. Dorneles, R. Gunning, P. Stamenov, J.M.D. Coey, Phys. Rev. B 74, 115307 (2006)
W.M. Yen, M.J. Weber, Inorganic phosphors. (CRC, Boca Raton, 2007)
M. Tamatani, in Phosphor handbook, ed. By W.M. Yen, S. Shionoya, H. Yamamoto. (CRC, Boca Raton, 2007), p. 167
K. Gopinadhan, S.C. Kashyap, D.K. Pandya, S. Chaudharya J. Appl. Phys. 102, 113513 (2007)
Z.M. Tian, S.L. Yuan, J.H. He, P. Li, S.Q. Zhang, C.H. Wang, Y.Q. Wang, S.Y. Yin, L. Liu, J. Alloys Compd. 466(1–2), 26 (2008)
A. Azam, A.S. Ahmed, M. Chaman, A.H. Naqvi, J. Appl. Phys. 108, 094329 (2010)
K. Vadivel, V. Arivazhagan, S. Rajesh, Int. J. Sci. Eng. Res. 2(4), 43 (2011)
N.S. Sabri, M.S.M. Deni, A. Zakaria, M.K. Talar, Phys. Procedia 25, 233–239 (2012)
M. Saravanakumar, S. Agilan, N. Muthukumarasamy, V. Rukkumani, A. Marusamy, A. Ranjitha, Acta Phys. Polonica A 127, 1656–1661 (2015)
V. Agrahari, A.K. Tripathi, M.C. Mathpal, A.C. Pandey, S.K. Mishra, R.K. Shukla, A. Agarwal, J. Mater. Sci. Mater. Electron. 26(12), 9571–9582 (2015)
N. Salah, S Habib, A Azam, M. Ansari, W.M. AL-Shawafi, Nanomater. Nanotechnol. 6, 17 (2016)
A.K. Singh, U.T. Nakate, Adv. Nanopart. 2, 66–70 (2013)
S. Singh, D. Gupta, V. Jain, A.K. Sharma, Mater. Manuf. Process. 30, 1 (2015)
L. Tan, L. Wang, Y. Wang, J. Nanomater. 2011, 1–10 (2011)
R.K. Mishra, P.P. Sahay, Ceram. Int. 38, 2295–2304 (2012)
W. Zhou, L. Liu, P. Wu, J. Magn. Magn. Mater. 321, 3356 (2009)
J. Tauc, R. Grigorovici, A. Vancu, Phys. Status Solidi B 15(2006), 627–637 (2006)
A. Sundaresan, R. Bhargavi, N. Rangarajan, U. Siddesh, C.N.R. Rao, Phys. Rev. B 74, 161306 (2006)
S Chacko, M.J. Bushiri, V.K. Vaidyan, J. Phys. D 39, 4540 (2006)
H. He, T.H. Wu, C.L. Hsin, K.M. Li, L.J. Chen, Y.L. Chueh, L.J. Chou, Z.L. Wang, Small 2(1), 116–120 (2006)
S. Luo, J. Fan, W. Liu, M. Zhang, Z. Song, C. Lin, X. Wu, P.K. Chu, Nanotechnology 17, 1695–1699 (2006)
S. Luo, P.K. Chu, W. Liu, M. Zhang, C. Lin, Appl. Phys. Lett. 88, 183112–183113 (2006)
K. Vanheusden, W.L. Warren, C.H. Seager, D.R. Tallant, J.A. Voigt, B.E. Gnade J. Appl. Phys. 79, 7983 (1996)
T. Krishnakumar, R. Jayaprakash, M. Parthibavarman, A.R. Phani, V.N. Singh, B.R. Metha, Mater. Lett. 63, 896 (2009)
P Zhan, W Wang, C Liu, Y. Hu, Z. Li, Z. Zhang, P. Zhang, B. Wang, X. Cao, J. Appl. Phys. 111, 033501 (2012)
J.D. Bryan, D.R. Gamelin Prog. Inorg. Chem. 54, 47–126 (2005)
A.K. Shukla, EMR/ESR/EPR spectroscopy for characterization of nanomaterials. (Springer, Technology & Engineering, New Delhi, 2016), p. 179
H.J. Zhou, D.M. Hofmann, A. Hofstaetter, B.K. Meyer, J. Appl. Phys. 94 1965 (2003)
P. Agoston, K. Albe, R.M. Nieminen, M.J. Puska, Phys. Rev. Lett. 103, 245501 (2009)
A. Kar, S. Kundu, A. Patra, J. Phys. Chem. C 115, 118–124 (2011)
N. Ozcan, T. Kortelainen, V. Golovanov, T.T. Rantala, J. Varra, Phys. Rev. B 81, 35202 (2010)
A. Jagannatha Reddy, M.K. Kokila, H. Nagabhushana, J.L. Rao, B.M. Nagabhushana, C. Shivakumara, R.P.S. Chakradhar, Spectrochimica Acta Part A 79, 476–480 (2011)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Thamarai Selvi, E., Meenakshi Sundar, S. Effect of Mn doping on structural, optical and magnetic properties of SnO2 nanoparticles by solvothermal processing. J Mater Sci: Mater Electron 28, 15021–15032 (2017). https://doi.org/10.1007/s10854-017-7376-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-7376-6