
Abstract
NiO nanoplates were prepared through a method base on cathodic electrochemical deposition-heat treatment. The deposition mechanism of the hydroxide precursor (i.e. Ni(OH)2) during the CED step, and final oxide formation during the HT step were specified. The structural and morphological evolutions by XRD, SEM and TEM confirmed the production of pure β-Ni(OH)2 nanoplates and cubic crystalline NiO nanoplates as the CED and HT products, respectively. The supercapacitive performance of the prepared NiO nanoplates was further studied by techniques of cyclic voltammetry and galvanostatic charge–discharge within the potential window of −0.1 to 0.5 V in 1 M KOH electrolyte. The measurements revealed that the nanoplates have high reversibility (ΔEp = 22 mV), high specific capacitance (1345 F g−1 at the scan rate of 2 mV s−1) and proper long-term cycling stability of 89.1 % capacity retention after 3000 cycling at the current density of 1 A g−1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The electrochemical double layer capacitors or EDLCs are classified into three categories: supercapacitor, ultracapacitor and pseudocapacitor [1]. Up now, various metal oxides like NiO [2, 3], Co3O4 [4, 5], MnO2 [6, 7], TiO2 [8] and RuO2 [9] have been used as electrode materials for capacitor applications. In cases where electrode of the EDLCs is made of metal oxides (such as nanostructured NiO), the resulting design makes use of two charge storage mechanisms, i.e. double layer capacitance and pseudo-capacitance; this design allows EDLC to achieve higher capacitance output, so this design is named accordingly as the supercapacitor (SC). Recently, the potential applications of nanostructured NiO have encouraged the researchers to pay further attention to the preparation of this material. These applications include the enhancement of supercapacitors [2–20], rechargeable alkaline batteries [21, 22], sensor [23–25], catalyst [26, 27], electrochromic films [28, 29] and solar cells [30]. This further attention has resulted in development of a number of nanostructured NiO synthesis methods, including surfactant template [31], hydrothermal [15–18, 32–34], solid state [35], thermal decomposition [36], solvothermal [28, 37, 38], chemical precipitation [2, 7, 39], and sol–gel [8, 40]. In addition, it has been found that the nanostructured NiO produced by electrochemical deposition (ED) methods including anodic [41–44] and cathodic [11, 12, 45–47] routes, can demonstrate better super-capacitive features. These superior supercapacitive features allow electrolytic NiO to be used more effectively in electrochemical supercapacitors. In support of this claim, it has been found that electrochemical performance of an electrolytic nanostructured NiO is far better than that of conventionally produced NiO [35]. The main challenge ahead of further use of nanostructured NiO however is the gap between the currently achieved specific capacitances which range from 100 to 1500 F g−1, and the theoretically achievable capacitance, which is about 3228 F g−1 at 0.4 V. This issue has encouraged the researchers to focus their efforts on developing new solutions for optimization of NiO microstructure in order to achieve better energy and power densities.
It has been reported that cathodic electrochemical deposition-heat treatment (CED-HT) is one of the effective methods for large scale synthesis of nanostructured metal oxides [46–52]. In this method, first metal hydroxide precursor is produced through CED step. Notably, base (OH−) electrogeneration on the cathode surface followed by chemical formation of hydroxide is the main mechanism during this step. Then, the final oxide is obtained by applying the HT procedure i.e. the hydroxide precursor is heat-treated at the proper atmosphere for the some hours. Despite its clear advantages, the use of this method for preparation of nanostructured NiO has been largely neglected [11, 12, 45–47], as researchers have been more focused on anodic variation of electrochemical synthesis of nanostructured NiO [41–44]. However, recently there have been some efforts to use this method for preparation of nano-structured NiO like capsule-like [11] and ultrafine nanoparticles [12].
This paper describes the process of producing uniform nanoplates of NiO. To produce NiO nanoplates, first the procedure of galvanostatic deposition was conducted with i = 0.1 mA cm−2 in a 0.005 M Ni (NO3)2 bath at a temperature of 60 °C, and then the resulting deposit was heat-treated for 3 h at a temperature of 400 °C. Examination of the resulting product by cyclic voltammetry (CV) and charge–discharge methods showed the excellent supercapacitive performance of the produced nanoplates.
2 Experimental procedure
2.1 Chemicals
Ni(NO3)2·6H2O (Merck), polytetrafluoroethylene (PTFE, Merck), acetylene black and KOH (Merck) were used as received. All solutions were prepared by using purified water by UHQ Elga System. Aqueous solution of 5 mM Ni(NO3)2·6H2O was prepared for electrodeposition.
2.2 Synthesis procedure
Deposition experiments were performed in the galvanostatic regime using a two electrode system. The electrochemical cell included a cathodic stainless-steel substrate centered between two parallel graphite anodes. Prior to each deposition, the steel substrates were given a galvanostatically electropolishing treatment [12]. Deposition experiments were carried out at the constant current density of 0.1 mA cm−2 for 1 h at bath temperature of 60 °C. After the deposition step, the steel electrode was taken out of the electrolyte and washed several time with deionized water, and finally dried at RT for 48 h. To obtain final oxide, the hydroxide deposit was then scraped from the steel electrode and heat-treated at 400 °C for 3 h.
2.3 Instrumentation
The crystal structure of the prepared samples was determined by powder X-ray diffraction (XRD, Phillips PW-1800) using Cu Kα radiation. These measurements were conducted in the diffraction angle (2θ) ranges of 5°–80° at 5° min−1. Thermogravimetric analysis (TGA) and differential scanning calorimetric (DSC) analysis tests were carried out in air, between room temperature and 600 °C at a heating rate of 5 °C min−1 using a thermoanalyzer (STA-1500). The morphology of the samples was examined using a scanning electron microscope (SEM, LEO 1455VP). Cyclic voltammetry (CV) and galvanostatic charge–discharge tests were performed using an Autolab workstation system (AUTOLAB®, Eco Chemie, PGSTAT 30). The volumetric N2 adsorption/desorption studies were performed, to analyzed surface area and pore structure of the prepared nanoplates, by Brunauer–Emmett–Teller (BET) measurement using at quanta-chrome NOVA-2200e surface area analyzer.
2.4 Electrochemical measurements
The electrochemical properties and capacitance of a supercapacitor electrode made of the prepared NiO nanoplates, were studied in a three-electrode system by CV and constant current charge and discharge tests, as well as EIS measurements. The working electrode was prepared by mixing the prepared NiO, conducting graphite and polytetrafluoroethylene (PTFE) binder (with the weight ratios of 85:10:5, respectively). The prepared mixture was pressed, under 10 MPa, into nickel foam current collectors (1 cm × 1 cm) and then dried in oven for 15 min at 60 °C. All experiments were carried out in a three compartment cell with a working electrode, a platinum plate (1 cm2) counter electrode and a An Ag/AgCl (1 M KCl, saturated) as the reference electrode. The electrolyte was 1 M KOH aqueous solution. CV tests were carried out between −0.1 and 0.5 V (vs. Ag/AgCl) at the different scan rates of 2, 5, 10, 25 and 50 mV s−1. The corresponding specific capacitance was calculated from the following equation:
where C is specific capacitance of NiO (F g−1) measured in the potential range of Va–Vc, m is the mass of NiO (g), v is the scan rate (V s−1) and I(V) is a current response depending on sweep voltage. The mass loading of active materials was 1.8 mg. Galvanostatic charge/discharge tests were performed in the potential range of −0.1 to 0.5 V (vs. Ag/AgCl) at the different current densities of 1, 2, 3, 5 and 10 A g−1. The SCs of the NiO electrode was calculated from the CP curves based on Eq. (2):
where C is the specific capacitance; where, I is the applied constant current (A), ΔV is the potential range (0.6 V), Δt is the time of a discharge cycle (s) and m is the mass of NiO (g).
3 Results and discussion
3.1 Hydroxide formation mechanism
The deposition of Ni(OH)2 on the steel surface during CED has been previously explained in Refs. [11, 12], and included the following stages:
-
Electrochemical stage:
$$2{\text{H}}_{2} {\text{O}} + 2{\text{e}}^{ - } \to {\text{H}}_{2} + 2{\text{OH}}^{ - } \quad {\text{E}}^\circ = - 1.08\,{\text{V versus Ag/AgCl}}$$(3) -
Chemical stage:
$${\text{Ni}}^{2 + } \left( {\text{aq}} \right) + 2{\text{OH}}^{ - } \left( {\text{aq}} \right) + x{\text{NO}}_{3}^{-} + y{\text{H}}_{2} {\text{O}} \to \left[ {{\text{Ni}}\left( {\text{OH}} \right)_{2 - x} \left( {{\text{NO}}_{3} } \right)_{x} \cdot z{\text{H}}_{2} {\text{O}}} \right] \downarrow$$(4)
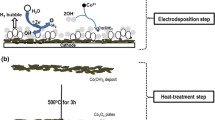
The potential value observed during the CED process (−1.01 V vs. Ag/AgCl) implicated that the H2O molecules reduction (Eq. 3) produces the base (OH−) ions at our applied parameters in CED of Ni(OH)2. Furthermore, gas bubbles releasing on the cathode surface fully confirmed the base production through H2O reduction. The formation of nickel hydroxide on the steel surface is schematically illustrated in Fig. 1. The electrochemical stage results H2O reduction and electrochemical generation of OH− (stage I in Fig. 1). The H2 gas bubbles were also generated on the cathode surface in this stage. By pH increasing to the required threshold value for hydroxide formation, nickel hydroxide is chemically formed and deposited on the steel cathode (stage II in Fig. 1).

Schematic view of NiO preparation by cathodic electrochemical deposition-heat treatment (CED-HT) method
3.2 Heat-treatment to obtain NiO
Nickel oxide was obtained via HT process at 400 °C for 3 h. The physico-chemical changes during HT were specified by thermogravimetric analysis and the results are shown in Fig. 2. The DSC curve of NiO nanoplates has four steps (i.e. A, B, C and D peaks) during the conversion of nickel hydroxide deposit to NiO. Further TG profile of nanoplates has the distinct weight losses as a result of these peaks. The first DSC peak (i.e. peak A) corresponded to the removal of H2O molecules attached into the surface of deposited hydroxide:

Thermogravimetric analysis of the deposited hydroxide precursor
The TG curve also has a relative sharp weight loss with 12.8 wt%. It has been reported that the hydroxide decomposition observes between 250 and 350 °C [12, 45–49]. Thus, step II (i.e. both B and C peaks) were assigned to the decomposition of Ni(OH)2 into NiO. In this step, water molecules are removed from the hydroxide plates through dehydroxylation:
For this step, a total weight loss of 18.2 wt% can be observed on the TG curve. Notably, the observed weight loss is rather less than the theoretically expected value (i.e. 19.49 wt% for the removal of one H2O molecule from Ni(OH)2). This indicates the decrease of structural water of the hydroxide sample as a result of the intercalation of nitrate ions.
The last broad DSC peak at step III (i.e. peak D) at 350–550 °C was attributed to the removal of nitrate from the structure [12, 49]:
TG curve also showed a 2.4 % weight loss within this temperature range and the overall weight loss for the sample was 33.4 %.
3.3 Structural and morphological characterization
3.3.1 Crystal structure
The XRD patterns of the products obtained from CED and HT steps are presented in Fig. 3. In Fig. 3a, the peaks corresponding to the crystal planes of (001), (100), (101), (102), (110) and (111) confirm the deposition of β-Ni(OH)2, as the hydroxide precursor (JCPDS 74-2075, solid lines in Fig. 3b). Figure 3b shows the XRD pattern of the oxide sample prepared after HT process. The observed XRD pattern is similar to that of crystalline cubic NiO structure (JCPDS 47-1049, solid lines in Fig. 3b). No diffraction peaks related to the Ni(OH)2 are also seen in the XRD pattern of oxide product, implicated that hydroxide precursor has been completely changed to NiO after HT at 400 °C.

XRD patterns of the samples obtained from a CED and b HT steps
3.3.2 Morphology
Morphology of the prepared samples through CED and HT steps is presented in Fig. 4. The SEM image (Fig. 4a) showed the plate-like structures for the hydroxide precursor obtained from CED step. In this image, it is observable that the hydroxide plates have grown in different directions on the steel during CED process, and also have non-stacked and free-standing form. This style of Ni(OH)2 plates may resulted from the effects of H2 bubbling during the electrosynthesis of hydroxide precursor. Notably, we previously found that the H2 bubbles could act as a dynamic temple for the deposit growth and directly effect on the morphology of the deposited metal hydroxide i.e. its growth direction, structure-type and nano-size. For example, we reported that H2 bubbling plays essential role in the formation of rod-, plate- and capsule-like nanostructures of metal hydroxides on the cathode surface during the cathodic electrosynthesis [50–58]. For the calcined sample, the complete plate morphology is clearly observable from the SEM and TEM images in Fig. 4b, c. The NiO sample has discrete plates and free-standing form at nanoscale, which can embody the prepared sample to have high surface area (as confirmed via BET analysis in Fig. 5). Furthermore, this style of NiO plates (i.e. their discrete form) can result the proper wettability of the fabricated electrode by electrolyte and enhance the charge storage ability. Notably, the edges of NiO plates are non-uniform which can be related to the physicochemical changes during HT step (i.e. removal of structural water from the hydroxide plates and their conversion to oxide plates).

SEM image of a electrodeposited precursor, b SEM and c TEM images of final oxide sample

BET surface area of NiO nanoplates obtained after HT step; a N2 adsorption–desorption isotherm and b BHJ pore size distribution
3.4 Surface area analysis
The surface charcateristics (surface area and pore size) of the prepared NiO plates were determined using the Brunauer–Emmett–Teller (BET) technique. The N2 desorption-adsorption curve and the corresponding BJH (Barret–Joyner–Halenda) pore size distribution profile of the NiO nanoplates are displayed in Fig. 5. The N2 isotherm of nanoplates shows irreversible type IV isotherm in the Brunauer classification, which is the typical feature of mesoporous materials according to the IUPAC classification [58]. Furthermore, the characteristic hysteresis loop observed in the P/Po range of 0.4–1 can be ascribed to H3 loop. The type of loop is usually observed in structures containing the plat-like particles assembled in slit-like pores [58]. Notably, type IV isotherm is also obtained with aggregates of plate-like particles, which possess non-rigid slit-shaped pores [19, 21], which is completely consist with SEM and TEM observations in Fig. 5. The H3 loop is connected with the secondary process of capillary condensation, suggesting the complete filling of the micropores [19, 21]. Because of delayed capillary condensation, multilayer adsorption is able to proceed on the surface of nanoplates until a high P/Po is reached. When condensation is occurred the state of the adsorbate changes and desorption curve follows a different path until the condensate becomes unstable at a critical P/Po [58]. As seen in Fig. 5a, this condensation is completely occurred at the pressure range of 0.5–0.97. The NiO nanoplates have a high BET surface area of 176 m2 g−1. The pore size distribution of the nanoplates was also analyzed through employing the BJH technique. From Fig. 5b, it was determined that the prepared sample has micropores with the average radius of ~1.5 to 1.7 nm. The high surface area and porous structure of the prepared nanoplates will allow better contact between electrolyte and NiO electrode for the transfer of electrons and ions (i.e. fast redox reactions), presumably leading to high energy storage ability.
As the charge storage mechanism of NiO nanoplates requires tunnel cavities for intercalation/extraction of H+ or OH− [58], it is expected that the these porous plates can facilitate the intercalation/extraction of H+ or OH−, since the sizes of these ions (3.58 Å) are smaller than the plate pore size cavities (15–17 Å).
3.5 Electrochemical evaluation
3.5.1 Cyclic voltammetry
The charge storage performance of the electrode prepared from NiO nanoplates was investigated by cyclic voltammetry (CV). Figure 6a indicates the CV of the prepared NiO electrode in 1 M KOH at the scan rate (SR) of 5 mV s−1. The one pair redox peak is observable in the CV profile of the prepared electrode as donated by P1 and P2 in Fig. 6a. In the anodic part, one anodic peak (P1) is located at ~0.44 V (vs. Ag/AgCl) prior to the oxygen evolution reaction (OER). Similarly, in the cathodic part, one cathodic peak (P2) is appeared at about 0.42 V during the reverse sweep. Notably, there is an important issue about the electrochemical behavior of the NiO electrode. In fact, as clearly observable on CVs, NiO electrode exhibits two different charge storage performance; (1) the electrical double-layer capacitance for potentials lower than 0.3 V, and (2) faradic storage for potentials higher than 0.3 V (as both behavior is clearly seen in Fig. 6a). Generally, the asymmetric redox peaks of CV profile indicate the irreversibility of the redox processes, inducing ohmic resistance and polarization during the redox reaction [17, 18]. In the case of our prepared electrode from NiO nanoplates, the CV curve was completely symmetric and so NiO nanoplates have low ohmic resistance. The ΔEp (Epa−Epc) is known as a readily measurable value for determination of the reversibility of the redox reaction. The nanoplates exhibited low a ΔEp value of 22 mV, which was very low as compared with those corresponding to NiO nanoflowers (ΔEp = 54 mV), nanoslices (ΔEp = 61 mV) and nanoparticles (ΔEp = 71 mV) reported in Ref. [40], and nano-porous NiO (ΔEp = 103 mV) reported in Ref. [58]. These observations confirmed an excellent reversibility of our prepared NiO nanoplates in cyclic voltammetry. The charge storage mechanism i.e. redox reactions of nickel oxide in aqueous KOH electrolyte has been previously discussed in detail [11, 12, 17–20]. Here, we briefly discuss on the charge storage procedure by NiO. Due to instability of NiO in the aqueous solution, a Ni(OH)2 layer is first formed at NiO surface [11]:

a CV of the NiO electrode at the scan rate of 5 mV s−1, b CVs at the different scan rates and c the calculated specific capacitances at the various scan rates
Then, the hydroxide to oxyhydroxide conversion process occurs as faradic redox reaction in aqueous KOH electrolyte:
It should be noted that hydroxyl ions and water molecules are not exist at the interface of Ni(OH)2/NiOOH. So, the Eq. (9) should be modified as [11, 59]:
Hence, it can be said that the anodic redox reaction of NiO electrode is occurred via the formation of NiOOH at its surface. Overall, the charge storage mechanism of the NiO nanoplates can be written as [33, 60]:
According to this mechanism, the surface area of NiO at nanoscale plays a key role in its behavior on the anodic part of CV curve. As confirmed by BET analysis in Fig. 5, the prepared nanoplates have high surface area which comforted the anodic reaction of NiO electrode (i.e. higher z value in Eq. 11) and so the high current density is seen on P1 peak at the anodic part of CV curve (Fig. 6b). The corresponding cathodic reaction of NiO electrode requires electrolyte penetration into the surfaces of nanoplates and H+ insertion from the electrolyte into the crystal lattice of NiO. In this case, the porous feature of the prepared nanoplates (as seen in BET analysis, Fig. 5b) facilitates the electrolyte penetration inside the NiO electrode (i.e. higher z value in Eq. 11) and enhanced the cathodic redox reaction (i.e. higher z value in Eq. 11), where the high current density is also observed for the P2 peak in the CV curve (Fig. 6b). Overall, the high surface area associated with porous texture of the prepared NiO nanoplates renders their CV curve as regular form i.e. the anodic charge is approximately equal to the cathodic one (Qa ≈ Qc). While in the case of capsule-like particles [11] and ultra-fine nanoparticles of NiO [12], although the large surface of these nanostructures could be facilitated the anodic part process (Q a), however, lack of porosity in these structures (as confirmed by SEM and BET) leads to lower penetration of electrolyte, reduction of the cathodic current (Q c) and unequally charge response of the anodic and cathodic process (where Q a > Q c). Accordingly, cyclic voltammetry curves obtained from the electrode fabricated using these nanostructures, exhibit irregular form. Therefore, in the case of electrochemical behavior of NiO, it can be said that, the high surface area of the prepared nanostructure is insufficient for the total reduction of NiO and exhibiting the better supercapacitive performance and the structure of the prepared electrode should be porous.
The CVs of the prepared nickel oxide electrode were recorded at various scan rates, which are shown in Fig. 6b. It can be seen that all CVs have uniform shapes, and increasing the scan rate has no change on the CV profile of the NiO electrode. This reveals the excellent redox reactions of the prepared electrode even at high scan rates. The average specific capacitance of the NiO was estimated from the CVs in Fig. 6b by calculating the area under i–V profile. Using Eq. (1), the specific capacitances of NiO nanoplates were determined to be 1505, 1345, 1176, 1057, 926, 744 and 554 F g−1 at the scan rates of 1, 2, 5, 10, 25, 50 and 100 mV s−1, respectively (Fig. 6c). The values showed excellent supercapacitive performance of the prepared NiO nanoplates. Notably, our prepared plates have some lack in the delivered specific capacitances at higher scan rates, where the calculated specific capacitances are unexpectedly reduced with increasing the scan rate. This charge storage defect has been also reported for pure NiO nanostructures [3–12]. This event can be related to the low accessibility of the electro-active materials at the high scan rates as compared with low applied scans [3, 11, 17]. At higher scan rates, the outer surfaces of the NiO plates are accessible and ionic diffusion takes place inside of this region, whereas at low scan rate, both inner and outer surfaces of plates have contribution in the charge storage and delivered specific capacitances. However, due to the lack of internal porosity of the prepared NiO plates the contribution of inner materials is reduced at the higher scan rates. Hence the calculated specific capacitances at the higher scan rates are somewhat reduced.
3.5.2 Charge–discharge tests
The galvanostatic charge and discharge (i.e. cycling) profiles for the prepared NiO electrode were recorded at the different current loads of 1, 2, 3, 5 and 10 A g−1, and the results are shown in Fig. 7a. All curves have symmetric triangular charge–discharge forms even at high current densities, suggesting excellent pseudo-capacitive behavior of NiO nanoplates, which is in agreement with CV analysis. The charging and discharging curves of the nanoplates are mostly symmetrical due to the kinetic reversibility of the hydroxyl ions on the porous NiO surface during the redox reaction [4, 12]. This is also supported by the similarity in the charging time (tC) and discharging time (tD). The specific capacitance of nanosheets was calculated using Eq. (2) and the results are shown in Fig. 7b. As can be seen in Fig. 7b, the prepared electrode was capable of delivering specific capacitances as high as 1526 F g−1 (0.2 A g−1), 1345 F g−1 (0.5 A g−1), 1206 F g−1 (1 A g−1), 1027 F g−1 (2 A g−1), 904 F g−1 (3 A g−1), 672 F g−1 (5 A g−1) and 545 F g−1 (10 A g−1), which are close to the calculated ones based on the CVs (Fig. 7b). The values also confirmed the excellent supercapacitive behaviour for the NiO nanoplates. Furthermore, the electrochemical performance of NiO nanoplates were comparable with those reported for other nanostructures; 372 F g−1 at 10 mV s−1 for Cu doped NiO [3], 470 F g−1 at 1 A g−1 for NiO nanoflakes [11], 1174 F g−1 at 2 A g−1 for NiO ultrafine nanoparticles [12], 617 F g−1 at 0.5 A g−1 for NiO/MWCNTs nanohybrid thin films [13], 734 F g−1 at 1 A g−1 nanosheet-assembled hollow NiO ball-flower [14], 322 F g−1 at 1 A g−1 for NiO/Ag nancomposite [15], 559 F g−1 at 5 mA cm−2 for La-doped NiO fibers [16], 274 F g−1 at 5 A g−1 for NiO nano-flakes/graphene nanocomposite [17], 619 F g−1 at 1 A g−1 for NiO microspheres [18], 351 F g−1 at 2 A g−1 for NiO nanowires [19], 950 F g−1 at 5 A g−1 for mesoporous NiO nanowalls grown on RGO [26], 148 F g−1 at 100 mV s−1 for nanostructured NiO films [35], 540 F g−1 at 2 A g−1 for NiO nanoflakes grown on porous graphene frameworks [36], 485 F g−1 at 1 A g−1 for NiO nanoflowers [40] and 351 F g−1 at 2 A g−1 for porous nickel oxide film [44].

a Charge–discharge profiles of NiO electrode and b calculated capacitances at the different current densities
The cycle life of the NiO nanoplates was studied via galvanostatic charge–discharge cycling (3000 cycles) in a 1 M KOH electrolyte. The charge–discharge profiles were obtained at the current densities of 1 and 5 A g−1. The capacity decay during cycling was calculated based on the obtained capacitance value for each cycle via Eq. (2), and the cycle life of the NiO electrode was then evaluated. Figure 8 shows the cycling performance of the NiO electrode during 3000 cycles at the discharge current densities of 1 and 5 A g−1. The specific capacitances of our electrode change from 1206 to 1000.6 F g−1 for applied discharge current of 1 A g−1, showing a 89.1 % capacity retention after 3000 cycling. Also, the NiO-based electrode exhibits the specific capacitance value of 446 F g−1 after 3000 discharge cycles, revealing 75 % capacity retention for cycling at 5 A g−1. The results showed that the prepared nanoplates have highly stable capacitance behavior during cycling and therefore can be suitable for long-time capacitor applications in KOH.

a Calculated specific capacitances and b capacity retention during 3000 charge–discharge cycling at the current densities of 1 and 5 A g−1
4 Conclusion
Well-oriented and uniform nanoplates of nickel oxide were obtained by heat treatment (HT) of Ni(OH)2 nanoplates prepared through cathodic electrochemical deposition (CED). The prepared NiO sample was evaluated by XRD, SEM, TEM and DSC-TG analyses. Furthermore, the electrochemical performance of the NiO nanoplates was determined by cyclic voltammetry and charge–discharge measurements. The formation mechanism the NiO nanoplates during HT step was defined by DSC-TG analyses. The electrochemical evaluations revealed that the NiO nanoplates display an excellent capacitive behavior with a specific capacitance of 1206 F g−1 at a current density of 1 A g−1 and a long-term retention of ca. 89 % after 3000 constant current cycling at the current load of 1 A g−1. The observed results established that the NiO nanoplates can be promising electroactive materials for supercapacitors.
References
V.C. Lokhande, A.C. Lokhande, C.D. Lokhande, J.H. Kim, T. Ji, J. Alloys Compd. 682, 381 (2016)
D.U. Lee, J. Fu, M.G. Park, H. Liu, A. Ghorbani Kashkooli, Z. Chen, Nano Lett. 16, 1794 (2016)
K. Sathishkumar, N. Shanmugam, N. Kannadasan, S. Cholan, G. Viruthagiri, J. Sol Gel. Sci. Technol. 74, 621 (2015)
M. Aghazadeh, R. Ahmadi, D. Gharailou, M.R. Ganjali, P. Norouzi, J. Mater. Sci. 27, 8623 (2016)
P. Razmjoo, B. Sabour, S. Dalvand, M. Aghazadeh, M.R. Ganjali, J. Electrochem. Soc. 161, D293 (2014)
R. Poonguzhali, N. Shanmugam, R. Gobi, A. Senthilkumar, G. Viruthagiri, N. Kannadasan, J. Power Sour. 293, 790 (2015)
M. Aghazadeh, M. Ghannadi Maragheh, M.R. Ganjali, P. Norouzi, D. Gharailou, F. Faridbod, J. Mater. Sci. 27, 7707 (2016)
P. Prasannalakshmi, N. Shanmugam, A. Senthil kumar, N. Kannadasan, J. Electroanal. Chem. 775, 356 (2016)
F.Z. Amir, V.H. Pham, D.W. Mullinax, J.H. Dickerson, Carbon 107, 338 (2016)
H. Xiao, F. Qu, X. Wu, Appl. Surf. Sci. 360, 8 (2016)
H. Mohammad Shiri, M. Aghazadeh, J. Electrochem. Soc. 159, E132 (2012)
A. Barani, M. Aghazadeh, M.R. Ganjali, B. Sabour, A.A. Malek Barmi, S. Dalvand, Mater. Sci. Semicond. Process. 23, 85 (2014)
G.S. Gund, D.P. Dubal, S.S. Shinde, C.D. Lokhande, A.C.S. Appl, Mater. Interfaces 6, 3176 (2014)
J. Wang, Y. Zhang, P. Wan, T. Li, D. Hou, S. Hussain, H. Shao, J. Mater. Sci. 27, 8020 (2016)
Y. Wang, Q. Su, J. Mater. Sci. 27, 4752 (2016)
J. Jia, F. Luo, C. Gao, C. Suo, X. Wang, H. Song, X. Hu, Ceram. Int. 40, 6973 (2014)
Y.G. Zhu, G.S. Cao, C.Y. Sun, J. Xie, S.Y. Liu, T.J. Zhu, X.B. Zhao, H.Y. Yang, RSC Adv. 3, 19409 (2013)
M. Fan, B. Ren, L. Yu, Q. Liu, J. Wang, D. Song, J. Liu, X. Jing, L. Liu, CrystEngComm 16, 10389 (2014)
X. Ren, C. Guo, L. Xu, T. Li, L. Hou, Y. Wei, A.C.S. Appl, Mater. Interfaces 7, 19930 (2015)
B. Li, M. Zheng, H. Xue, H. Pang, Inorg. Chem. Front. 3, 175 (2016)
F. Zheng, S. Xu, Y. Zhang, J. Mater. Sci. 27, 3576 (2016)
Z. Wang, M. Zhang, J. Zhou, A.C.S. Appl, Mater. Interfaces 8, 11507 (2016)
Q. Gao, W. Zeng, R. Miao, J. Mater. Sci. 51, 2310 (2016)
Y. Zhang, L. Xie, C. Yuan, C.L. Zhang, S. Liu, Y. Peng, H. Li, M. Zhang, J. Mater. Sci. 27, 1817 (2016)
T. Geng, X. Zhou, H. Wu, J. Mater. Sci. 27, 8416 (2016)
S.R. Yousefi, D. Ghanbari, M. Salavati-Niasari, M. Hassanpour, J. Mater. Sci. 27, 1244 (2016)
F. Yu, X. Xu, H. Peng, H. Yu, Y. Dai, W. Liu, J. Ying, Q. Sun, X. Wang, Appl. Catal. A 507, 109 (2015)
M.M. Gomaa, M. Boshta, B.S. Farag, M.B.S. Osman, J. Mater. Sci. 27, 711 (2016)
R. Sharma, A.D. Acharya, S. Moghe, S.B. Shrivastava, M. Gangrade, T. Shripathi, V. Ganesan, Mater. Sci. Semicond. Process. 23, 42 (2014)
T. Okumura, T. Sugiyo, T. Inoue, M. Ikegami, T. Miyasaka, J. Electrochem. Soc. 160, H155 (2013)
P.M. Ponnusamy, S. Agilan, N. Muthukumarasamy, M. Raja, D. Velauthapillai, J. Mater. Sci. 27, 399 (2016)
A.K. Ramasami, M.V. Reddy, G.R. Balakrishna, Mater. Sci. Semicond. Process. 40, 194 (2015)
H. Pang, Q. Lu, Y. Zhang, Y. Li, F. Gao, Nanoscale 2, 920 (2010)
Y. Jiang, X. Leng, Z. Jia, H. Chen, H. Suo, C. Zhao, J. Mater. Sci. 26, 2995 (2015)
M. Ranjbar, M.A. Taher, A. Sam, J. Mater. Sci. 26, 8029 (2015)
K. Motevalli, Z. Zarghami, M. Panahi-Kalamuei, J. Mater. Sci. 27, 4794 (2016)
Y. Zhang, Y. Gui, X. Wu, H. Feng, A. Zhang, L. Wang, T. Xia, Int. J. Hydrogen Energy 34, 2467 (2009)
J.H. Pan, Q. Huang, Z.Y. Koh, D. Neo, X.Z. Wang, Q. Wang, A.C.S. Appl, Mater. Interfaces 5, 6292 (2013)
V. Rajendran, K. Anandan, Mater. Sci. Semicond. Process. 38, 203 (2015)
S.I. Kim, J.S. Lee, H.J. Ahn, H.K. Song, J.H. Jang, A.C.S. Appl, Mater. Interfaces 5, 1596 (2013)
M.S. Wu, Y.A. Huang, C.H. Yang, J.J. Jow, Int. J. Hydrogen Energy 32, 4153 (2007)
M.S. Wu, Y.A. Huang, J.J. Jow, W.D. Yang, C.Y. Hsieh, H.M. Tsai, Int. J. Hydrogen Energy 33, 2921 (2008)
M.S. Wu, Y.A. Huang, C.H. Yang, J. Electrochem. Soc. 155, A798 (2008)
M.S. Wu, M.J. Wang, J.J. Jow, J. Power Sour. 195, 3950 (2010)
K.R. Prasad, N. Miura, App. Phys. Lett. 85, 4199 (2004)
M. Wu, J. Gao, S. Zhang, A. Chen, J. Power. Sour. 159, 365 (2006)
V. Srinivasan, J.W. Weidner, J. Electrochem. Soc. 147, 880 (2000)
K. Sathishkumar, N. Shanmugam, N. Kannadasan, S. Cholan, G. Viruthagiri, Mater. Res. Express 1, 026104 (2014)
M. Aghazadeh, M.G. Maragheh, M.R. Ganjali, P. Norouzi, F. Faridbod, Appl. Surf. Sci. 364, 141 (2016)
M. Aghazadeh, M. Hosseinifard, Ceram. Int. 39, 4427 (2013)
M. Aghazadeh, M. Ghaemi, A.N. Golikand, A. Ahmadi, Mater. Lett. 65, 2545 (2011)
T. Yousefi, M. Aghazadeh, A.N. Golikand, M.H. Mashadizadeh, Sci. Adv. Mater. 4, 214 (2012)
M. Aghazadeh, S. Dalvand, J. Electrochem. Soc. 161, D18 (2014)
M. Aghazadeh, M. Hosseinifard, B. Sabour, S. Dalvand, Appl. Surf. Sci. 287, 187 (2013)
F. Khosrow-pour, M. Aghazadeh, B. Arhami, J. Electrochem. Soc. 160, D150 (2013)
I. Karimzadeh, M. Aghazadeh, B. Safibonab, M.R. Ganjali, S. Dalvand, Russ. J. Electrochem. 51, 263 (2015)
F. Rouquerol, J. Rouquerol, K. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications (Academic Press, San Diego, 1999)
S. Kumar Meher, P. Justin, G. Ranga Rao, Nanoscale 3, 683 (2011)
S.K. Meher, P. Justin, G. Ranga Rao, Electrochim. Acta 55, 8388 (2010)
M. Wohlfahrt-Mehrens, R. Oesten, P. Wilde, R.A. Huggins, Solid State Ionics 86, 841 (1996)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Aghazadeh, M. Synthesis, characterization, and study of the supercapacitive performance of NiO nanoplates prepared by the cathodic electrochemical deposition-heat treatment (CED-HT) method. J Mater Sci: Mater Electron 28, 3108–3117 (2017). https://doi.org/10.1007/s10854-016-5899-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-016-5899-x