
Abstract
The structural, optical and magnetic properties of pure and Sm doped SnO2 nanoparticles synthesized in aqueous solution by a low cost chemical co-precipitation method without using any stabilizing agent have been reported. X-ray diffraction analysis reveals that the crystallite size of SnO2 decreases with the increase of Sm doping into the SnO2 matrix. The calculated value of optical band gap of undoped SnO2 nanoparticles is calculated to be 2.65 eV and it anomalously increases with the increase of Sm3+ concentration. The band gap of undoped SnO2 nanoparticles is found to be lower than those reported for bulk SnO2 (3.6 eV) that may be due to the presence of defects, oxygen vacancies and non-stoichiometry of SnO2 nanoparticles. All the undoped and Sm doped SnO2 nanoparticles show weak room temperature magnetism in the range of applied low magnetic field region of 2000 Oe due to the presence of defects and oxygen vacancies. All the samples exhibit negative magnetic susceptibility for the applied magnetic field higher than 2000 Oe. The strong optical absorption with weak magnetic properties at room temperature may serve the SnO2 nanomaterials as a potential candidate for many DMS, spintronics, and optoelectronics based applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The semiconducting oxide nanomaterials are the hot area of research interest for scientific and technological advancement in the field of nanoelectronics, optoelectronics, photovoltaic’s, magneto-optics, magneto-electronics and photonic devices. Recently metal oxide based semiconductors such as TiO2, ZnO, SnO2, In2O3, CdO, ZrO2 and WO3 etc. are popular materials to investigate their unique properties at nanoscale, which are different from those of their corresponding bulk state [1–5]. Among various metal oxides, SnO2 is one of the widely studied n-type wide band gap semiconductor (Eg = 3.6 eV, at 330 K, unit cell parameters are a = 4.737 Å and c = 3.185 Å and its space group is P42/mmm). The high optical transparency, chemical and electrochemical stability, good electrocatalytic activity, nontoxicity and plentiful oxygen vacancies are the key factors for its potential applications in the transparent conducting electrodes [6], gas sensors [7], Li batteries, photoactive systems, liquid crystal display transistors, ferroelectric transparent thin film transistors, catalysts, optoelectronic devices and spintronic devices [8]. The effect of various doping elements like Fe, Ti, Cr, In, Cu, Rh, Co, Pb, Pd, Er, Mn, Sb, Ta, F, Sm, N and Ni etc. have been studied by many researchers to tailor the energy band gaps, structural, electrical, magnetic, electronic, electrochemical, optical and optoelectronic properties of SnO2 based nanomaterials [9–19]. In spite of the many work researches on doped SnO2 nanostructures, the rare earth (RE) ions doped SnO2 based nanostructures are still the subject of in-depth study for band gap alignments, site symmetries, magnetic and luminescence properties. Amongst the lanthanides, samarium (Sm) is a very active member having interesting properties that has tremendous applications in high-density optical storage, under sea communication and colour displays and its emitting 4G5/2 level exhibits relatively high quantum efficiency due to its fluorescence property [9]. The effect of Sm doping into the SnO2 host matrix has not been rigorously studied so far for its various applications in electronic, photonic, optical and optoelectronic devices. In this direction, Sm3+ ions are already reported to be most interesting lanthanide ions for its multiple energy-level structure and high fluorescence efficiency, which act as a great emitting centres and a spectroscopic probe due to presence of hypersensitive transitions to the symmetry of the ion environment [20]. It is essential to extend the study of Yanes et al. [20] to investigate rigorously the other structural, electronic, optical and magnetic properties of Sm3+ doped SnO2 for its possible applications in future optoelectronic devices. The authors [20] have used sol–gel method to synthesize the materials for studying its structural and morphological and emission properties. In the present study the most promising co-precipitation method has been used because it provides the control over the particle size, morphology, and surface chemistry by controlling the processing parameters such as solution composition, pH, reaction temperature, reaction pressure, ageing time, and nature of solvent involved in the process [21]. The present work reports the systematic study of Sm3+ doped SnO2 nanostructures to explore its structural, optoelectronics and magnetic properties by the help of various characterizations techniques such as X-ray diffraction (XRD), thermogravimetry analysis (TGA), transmission electron microscopy (TEM), scanning electron microscopy (SEM), vibration sample magnetometer (VSM), UV–visible spectroscopy and photoluminescence (PL) spectroscopy.
2 Experimental details
2.1 Synthesis
The approach for synthesizing the pure SnO2 and Samarium doped SnO2 nanoparticles is simple, solution-based chemical co-precipitation method which is inexpensive as compared to commercially available other techniques. All the chemical reagents used were of AR grade, and used directly without further purification. In typical synthesis of SnO2 the stannic tetrachloride hydrated (SnCl4·5H2O), 3.5058 gm was dissolved in 100 mL distilled water to prepare 0.1 M solution under vigorous magnetic stirring for 30 min at room temperature. 3 mL of an aqueous ammonia solution was added to the above solution drop wise under stirring to maintained pH in between 7 and 7.5. The dropping rate was well controlled for the chemical homogeneity and to form white slurry. After 18 h of ageing the slurry was filtered and washed for 8 h with distill water to remove impurities from the precipitate. The resulting precipitate was dried in an oven at 80 °C for 10 h to remove water molecules and then calcinated at 400 °C for 2 h in a furnace and the calcinations process started at room temperature. After calcinations the samples were crushed into fine powder to get undoped tin oxide (SnO2) nanomaterials.
For the synthesis of Sm doped SnO2, the stannic tetrachloride hydrated (SnCl4·5H2O), 3.5058 gm and samarium (III) nitrate hexahydrate Sm(NO3)3·6H2O, 0.0444 gm (1 at%) was dissolved in 100 mL distilled water to prepare 0.1 M solution under vigorous magnetic stirring for 30 min. An aqueous ammonia solution was added to the above solution drop wise under stirring and to maintain the pH in between 7 and 7.5. This process was repeated for 3 at% (0.1333 gm), 5 at% (0.2223 gm), 7 at% (0.3112 gm) and 10 at% (0.4446 gm) of Sm doping. After 18 h of ageing the slurry was filtered and washed for 8 h with distilled water to remove impurities from the precipitate. This precipitate was then dried in an oven at 80 °C for 10 h to remove water molecules and then calcinated at 400 °C for 2 h in a furnace. After calcinations the samples were crushed into fine powder to get Sm doped tin oxide nanomaterials of different doping concentration. Figure 1 shows the flow chart for the synthesis of Sm doped SnO2 nanoparticles.

Flow chart for synthesis of Sm doped SnO2 nanoparticles by co-precipitation method
2.2 Instrumentation
The structural properties of synthesized undoped and Sm doped SnO2 powder samples were analysed by XRD measurements using powder diffractometer (Bruker AXS D8) with Cu–Kα radiation (λ = 1.5406 Å). The size and shape of the synthesized powder were studied by TEM (Hitachi- H-8100) measurements. The FT-IR spectra of the synthesized samples have been recorded in the spectral range of 500–4000 cm−1 with Perkin–Elmer 1600 Fourier transform instrument. KBr was mixed in powder samples and then pelletized to record the FTIR spectra. The UV–Vis absorption spectra of all the samples were taken after ultrasonication of a small amount of sample dissolved in distilled water in the spectral range of 275–800 nm by Perkin–Elmer Lambda 35 UV–Vis spectrophotometer. The room temperature Raman spectroscope (RS) of the powder samples were recorded in the range of 200–1000 cm−1 using thermo scientific DXR—XI Raman imaging microscope. The 532 nm laser line of the Ar+ ions laser was used to illuminate the powder samples. Thermal properties of the synthesized samples were measured by thermo gravimetric analyser measurements using (TA Instruments Q50). The photoluminescence emission spectra of the synthesized samples were recorded by using a spectro-fluorometer (Spex Flurolog 3, FL3-22) with a 450 W Xenon Lamp as the excitation source with 330 nm excitation wavelength. For recording the PL spectra, emission slit width was kept to 3.0 nm. The room temperature magnetic properties of the synthesized samples were measured by a vibrating sample magnetometer (VSM) (Microsense EV7).
3 Results and discussion
3.1 X-ray diffraction
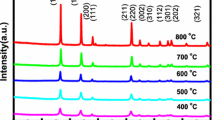
The typical XRD patterns of the undoped and Sm doped (1, 3, 5, 7 and 10 at%) SnO2 samples calcined at 400 °C are shown in Fig. 2. The peak positions of each sample exhibit the rutile phase tetragonal structure of SnO2, as confirmed from the ICDD card No. 77-0452 and JCPDS card No. 77-045 [5, 21]. The broadening in (110) and (101) peaks increases with the increase in Sm doping into SnO2 matrix. As the Sm concentration increases the crystallinity and average crystallite size of doped SnO2 decreases with respect to undoped SnO2 sample. The average crystalline size was estimated using Scherrer’s formula for the tetragonal phase structure of the samples from most intense XRD peaks and is presented in Table 1 [5]. Further, No other impurity peak related to Sm dopant was observed in the XRD pattern within the limits of instrumental sensitivity which shows the single phase SnO2 formation. When the particle size decreases to the nanometer scale, no crystal lattice of a long-range order exists in the SnO2 nanoparticles. Thus, the lack of its long-range order causes a reduction in the intensity of the peaks in the XRD pattern. Further, it is not possible for Sm+3 (0.109 nm) to occupy the regular lattice site of Sn4+ in SnO2 matrix due to its larger ionic radius than that of Sn+4 (0.069 nm) [22]. Thus, Sm creates defects and distortion in the SnO2 lattice that reduces the crystallinity of SnO2 and results into noise in the XRD pattern of Sm doped SnO2 samples.

XRD pattern of undoped and Sm doped SnO2 nanoparticles calcined at 400 °C
It means that even though the Sm ions does not occupy the regular lattice site of Sn4+, but it produces crystal defects and lattice mismatching around the dopants resulting into the charge imbalances that are responsible to change the stoichiometry of the host materials [1]. The lattice constants ‘a’ and ‘c’ have been determined by the following relation [3, 23] and are presented in Table 1:
where ‘d’ is the interplaner spacing and (h, k, l) are Miller indices, respectively. By looking at Table 1, it is clear that lattice parameters are significantly affected by Sm doping into SnO2 matrix.
3.2 Thermogravimetry analysis
TGA profiles of undoped and Sm doped SnO2 samples are depicted in Fig. 3. The weight loss in the temperature range between 90–200 °C can be attributed to the loss of adsorbed water in the tin oxide [24]. The combustion of residual organic species, including the decomposition of Cl ions are responsible for the weight loss between 300 and 550 °C [24].

TGA of different Sm doped SnO2 nanoparticles calcined at 400 °C
In Fig. 3, the maximum weight loss is 5 % for 10 at% Sm dopant concentration while the weight loss is minimum (2.5 %) for undoped SnO2. This weight loss increases with the increase of Sm content which indicates the increase of adsorbed water and higher residual organic species in the samples.
3.3 FTIR spectra
FTIR spectra of undoped and Sm doped (1, 3, 5, 7 and 10 at%) SnO2 nanoparticles recorded at room temperature in the range of 500–4000 cm−1 is presented in Fig. 4. It is evident from spectra, that undoped and Sm doped SnO2 nanoparticles still contain the water molecules since H2O molecules (bending vibrations in the range of 1500–1650 cm−1) have the property to be chemisorbed easily on the SnO2 surface when exposed to the atmosphere [9]. The broad spectra centred at ~3745 cm−1 is assigned to the stretching vibrations of O–H. The presence of these two bands indicates that a large amount of hydroxyl group are associated with the SnO2 surface in both undoped and Sm doped samples.

FTIR spectra of different Sm doped SnO2 nanoparticles calcined at 400 °C
The weight loss corresponding to H2O molecule and hydroxyl group is well supported by TGA also. The peaks at the low wavenumbers (500–1000 cm−1) may be attributed to the characteristic peaks of SnO2. The band appeared at ~697 cm−1 is assigned to the Sn–O antisymmetric vibrations [9]. The band observed at ~2893 cm−1 is assigned to C–H stretching mode of vibrations for the samples [5, 9]. An additional peak observed at around 2358 cm−1 is observed which shows the existence of CO2 (which usually appear around 2361 cm−1) molecules in air.
3.4 Raman spectra
Figure 5 shows the Raman spectra for undoped and Sm doped SnO2 nanoparticles calcined at 400 °C. The peak positions corresponding to various Raman bands of rutile SnO2 which belongs to the point group were indexed in the spectra. SnO2 having tetragonal rutile crystalline structure consists of 6 unit cell atoms which give a total of 18 branches for the vibration modes in the first Brillouin’s zone. According to group theory, the normal lattice vibrations at the gamma point of the Brillouin zone are Γ = 1A1g + 1A2g + 1A2u + 1B1g + 1B2g + 2B1u + 1Eg + 3Eu, where the B1g, Eg, A1g, and B2g modes of SnO2 are Raman active, A2u and Eu are infrared (IR) active, and A2g and B1u are inactive in both Raman and IR [5, 25]. The Raman bands of the pure rutile phase of SnO2 and Sm doped SnO2 were observed in different range. The main dispersion peaks in the range of 470–595, 623–633, 766–779 cm−1 were assigned for the three allowed fundamental modes Eg, A1g, and B2g, respectively of rutile SnO2 vibrations. The A1g and B2g modes are non degenerate and might be related to the expansion and contraction of the vibrating mode of Sn–O bonds, whereas the doubly degenerate Eg mode may be related to the vibrations of oxygen in the oxygen plane. The A1g and B2g modes vibrate in the plane perpendicular to the c-axis, whereas the Eg mode vibrates in the direction of the c-axis. In addition, few weak Raman bands are located for all the samples in the range of 300–320 cm−1 that are associated with the surface modes of SnO2 nanostructures [5, 25–27]. Xi and Ye [28] have also observed few new additional Raman modes for ultrathin SnO2 nanorods as compared to bulk SnO2, which were related to the facet surface area of a crystal, with small grain size of nanoscale SnO2. Beside this, the Raman spectra were compared with Raman active modes as predicted by group theory for B-type Sm2O3 structures. Anandhan et al. observed different lines at 457, 335, 152 cm−1 which were assigned to Ag modes and 424 cm−1 to both Bg and Ag modes for Sm2O3 structures [27]. Therefore, in this present work there is no mode of vibration mode for samarium (Sm3+) based oxide nanostructures (Sm2O3) which are in good agreement with the XRD results. As the Sm dopant concentration increases, the new weak bands arise in the range of 247–300 cm−1. This behaviour might be due to the fact that the addition of Sm might be is responsible for the changes in local disorder and defects such as vacant lattice sites or vacancy cluster or clusteration which may result in lattice distortion or reduction in lattice space symmetry. In case of all the samples, the peak positions drastically shifts either towards a higher wave number or lower wave number depending on the Sm doping concentrations. Similar anomalous shifting in the Raman spectra has been previously reported for TiO2 nanostructures with respect to the changes in annealing temperature [29].

Raman spectra of different Sm doped SnO2 nanoparticles calcined at 400 °C
3.5 Morphology analysis
The shape and size of the nanoparticles has a direct relationship with the crystal symmetry, growth methods, and growth conditions (such as volume, pressure and temperature etc.). Mainly, two quantities (1) chemical potential (2) surface energy for the atoms at surfaces play an important role for structuring the shape and size of the nanomaterials. The SEM images recorded for undoped and Sm doped SnO2 samples are shown in Figs. 6a–d and 7a–d at different scale of magnification.

SEM micrographs at 5 μm scale, a undoped SnO2; b 1 at% Sm doped SnO2; c 5 at% Sm doped SnO2; d 10 at% Sm doped SnO2 nanoparticles

SEM micrographs at 10 μm scale, a undoped SnO2; b 1 at% Sm doped SnO2; c 5 at% Sm doped SnO2; d 10 at% Sm doped SnO2 nanoparticles
In the SEM micrographs, it is clearly observed that the grain size and agglomeration remains similar with the increase in doping concentration of Sm precursor in SnO2 matrix. It means that the samarium doping into SnO2 does not lead to an extra clusteration. TEM micrographs recorded for undoped SnO2 are shown in Fig. 8a–c, while for 5 at% Sm doped SnO2 nanoparticles are presented in Fig. 9a–d, respectively. The undoped SnO2 nanoparticles show well isolated spherically shape, while 5 at% Sm doped SnO2 nanoparticles are not exactly spherical in shape. In fact the granular form of spherical nanoparticles are observed. It is observed that as the dopants precursor was added to SnO2 precursor, the average particle size decreases. The average particle size as estimated from particle size distribution curve (Figs. 8c, 9c) is calculated to be 7.5 ± 1.6 and 7 ± 2 nm for undoped and 5 at% Sm doped SnO2 nanoparticles, respectively.

TEM micrographs of SnO2 nanoparticles, a SnO2 at 50 nm scale; b SAED pattern and c particle size distribution curve

TEM micrographs of 5 at% Sm doped SnO2 nanoparticles, a SnO2 at 50 nm scale; b SAED pattern; c particle size distribution curve and d EDX pattern
The results are consistent with the crystallite size calculated from the XRD patterns. The selected area electron diffraction (SAED) patterns for both the undoped and Sm doped SnO2 indicates polycrystalline nature of the samples. All the main identified planes such as (110), (101), (211) are indexed in SAED pattern, which are in good agreement with the results obtained from XRD patterns. The elemental analysis of the Sm-doped SnO2 has been carried out for one of the samples and it is shown in Fig. 9d. The impurity peaks have been not observed in the EDX pattern of doped samples. From the EDX graph of 5 at% Sm doped SnO2, the peaks corresponding to Sm, Sn and O can be easily seen that further confirms the presence of Sm ions into the host SnO2 matrix.
3.6 UV–visible spectroscopy
UV–visible absorption spectroscopy is used to explore the optical properties of pure and Sm doped SnO2 (1, 3, 5, 7 and 10 at%) nanoparticles calcined at 400 °C. The absorbance is expected to depend on several factors, such as band gap, oxygen deficiency, surface roughness and impurity centres. The optical absorption spectra of undoped and Sm doped (1, 3, 5, 7 and 10 at%) SnO2 nanoparticles are shown in Fig. 10.

UV–Vis absorption spectra of undoped and Sm doped SnO2 nanoparticles
The optical band gap energy of the corresponding samples is calculated from Tauc plot by extrapolating the linear portion of (αhν)2 versus hν plots to intercept the photon energy axis as shown in Fig. 11 by using the following equation [24, 29].

(αhν)2 versus hν plots for undoped and Sm doped SnO2 nanoparticles
Where hν is the photon energy, α is the absorption coefficient, B is a constant relative to the materials and n is a value that depends on the nature of transition (n = 2 for direct band gap, 2/3 for direct forbidden gap and 1/2 for indirect band gap). The calculated band gap is 2.65, 3.16, 2.82, 2.78, 2.91 and 2.75 eV for undoped SnO2, 1, 3, 5, 7 and 10 at% Sm doped SnO2 nanostructures, respectively. In thise case the bandgap of undoped SnO2 bandgap (2.65 eV) is quite low as compared to the bandgap of bulk SnO2 (3.6 eV). This reduction in the band gap gives an indication of the stoichiometry deviation and degeneracy of the doped SnO2 and the increase in oxygen vacancies within the lattice. The results are consistent with those studied by Drake and Seal [15] for the band gap energy modifications in indium (In3+) substituted SnO2 nanocrystallines. The possible explanation for the ability of a material to decrease its band gap energy is that the density of surface states induced by chemisorbed oxygen species decreases with decreasing particle size, which would lead to a lesser degree of Fermi pinning. This allows the surface barrier to undergo larger changes and control the ability of charged species to move through the material. This decrease in density of surface states is directly caused by the curvature of the nanoparticles. A combination of the decrease in the density of surface states and the large amount of oxygen vacancies retained in the material explains the departure in the band gap values from the bandgap of bulk SnO2 (3.6 eV) [15]. The increase in bandgap after Sm doping is anomalous as there is no clear trend either for increase or decrease in the band gap for Sm doped SnO2. The results are quite interesting as the band gap for Sm doped SnO2 is found to be larger than the undoped SnO2 which indicates the blue shift in doped sample as compared to SnO2. The blue shift in the present study cannot be considered due to quantum-confinement effect as the atomic Bohr radius (~2.2 nm for SnO2) is very less than the radius of all the Sm doped SnO2 (~3.5 nm for 5 at% Sm doped SnO2) nanostructures. This anomalous behaviour in the bandgap may be influenced both by the oxygen concentration and intrinsic differences (like native defects and strain) in the electronic structure of the different samples as a function of doping concentration.
3.7 Photoluminescence
The room-temperature PL spectra of undoped and Sm (1, 3, 5, 7 and 10 at%) doped SnO2 nanoparticles under excitation at the wavelength of 330 nm are depicted in Fig. 12. A strong emission peak around ~393 nm (3.1 eV) can be observed from the spectra for undoped and Sm doped SnO2 nanoparticles which is comparable to the band edge transition energy of SnO2 nanoparticles. The emission band at ~393 nm is attributed to the direct recombination of a conduction electron in the Sn 4p band and a hole in the O 2p valence band [30]. The PL spectra of prepared samples are obtained as a result of the electron–hole separations, electron–phonon scattering and electron–hole recombination. Generally, oxygen vacancies are known to be the most common defects in semiconductor nanocrystals and usually act as radiative centre of luminescence in visible region. In SnO2, oxygen vacancies are present in three different charge states: \({\text{V}}_{\text{o}}^{0}\), \({\text{V}}_{\text{o}}^{ + }\) and \({\text{V}}_{\text{o}}^{2 + }\), in which \({\text{V}}_{\text{o}}^{0}\) is very shallow donor. The origin of visible emission can be assigned to the recombination of electrons in the shallow levels with the photo excited holes in the valence band [1]. Interestingly in the PL spectra of all the samples a broad emission edge is observed in the range of 400–500 nm which is the indication for the presence of multiple peaks under a main peak corresponding to multiple emissions from oxygen vacancies present in the samples. These vacancies give rise to the defect levels in the crystal and enhance the visible emission in the spectra [1]. From PL spectra of the Sm doped SnO2 nanoparticles, it is also observed that emission peaks shifts towards lower wavelength side (Blue shift) with respect to undoped SnO2 which is consistent with the results obtained from UV results spectra. This shifting in doped tin oxide nanoparticles may be attributed to the increase in band gap with the increase of Sm content in SnO2.

Photoluminescence spectra (PL) of undoped and Sm doped SnO2 nanoparticles
3.8 Magnetic properties
The field dependent magnetization (M–H curve at 300 K) measurement of undoped and Sm (1, 5 and 10 at%) doped SnO2 nanocrystalline samples calcined at 400 °C are shown in Fig. 13. The magnified region of the graph (in the region −2000 to 2000 Oe) is shown in Fig. 13b. All measurements have been corrected by eliminating the signals from the sample holder. All curves clearly exhibit hysteresis behaviour with coercivity and magnetization under a strong externally applied magnetic field of 20 kOe. The graph exhibits weak ferromagnetism in nature at room temperature for undoped and doped SnO2 nanoparticles under a weak applied magnetic field region of 2000 Oe. In general the week ferromagnetism is not expected in undoped doped and defects free SnO2 due to the nonexistence of unpaired electrons. There are several reports for room temperature (RT) ferromagnetism in doped SnO2 [5, 31–33]. The weak ferromagnetism in undoped and doped SnO2 at lower applied magnetic field is attributed to the oxygen vacancies (Vo) and the surface defects created during synthesis process [33, 34]. The presence of oxygen vacancy defects are already confirmed by PL spectra. The maximum coercivity (~324 Oe) was observed for 10 at% Sm doped SnO2 nanocrystalline sample. As the strength of externaly applied field is increased, the magnetic susceptibility dramatically goes negative and consequently the graphs show diamagnetic nature (diamagnetic above 2000 Oe for undoped SnO2 and 6500 Oe for 10 at% Sm doped SnO2) for the samples.

a M–H curve of undoped and Sm doped (1, 5 and 10 at%) SnO2 nanoparticles and b magnified graph
The diamagnetic nature with a linear negative behaviour which increases as the magnetic field increases is assigned to the diamagnetic contribution coming from the sample holders. Therefore no saturation magnetization has been observed for Sm doped SnO2 nanoparticles. The first principles calculations by Wang et al. [31] revealed that s and p electrons of oxygen atoms in SnO2 can be spin polarized, resulting in net magnetic moment for pure SnO2 nanosheets. There are several mechanisms proposed in the literature to explain the ferromagnetism and its origin in diluted magnetic semiconductors (DMS) but the exact mechanism is still under debate [5, 31–37]. More studies are needed to understand such room temperature magnetic behaviour of SnO2 based DMS nanostructures.
4 Conclusions
Undoped and Sm doped SnO2 nanoparticles were synthesized by a simple chemical co-precipitation method. The tetragonal phase of undoped and doped SnO2 nanoparticles was confirmed from XRD and SAED analyses. The undoped SnO2 nanoparticles are spherical in shape while the doped SnO2 nanoparticles show granular shape of partially spherical nanoparticles. TEM analysis revealed that particle size decreased with Sm doping into SnO2 matrix. The calculated optical band gap of undoped SnO2 nanoparticles is 2.65 eV and anomalously increases with the doping of Sm into SnO2 matrix. Room temperature ferromagnetism was found in undoped and Sm doped SnO2 nanoparticles at in the lower region of applied magnetic field which drastically changes to negative susceptibility with diamagnetic behaviour at higher (>2000 Oe) magnetic field.
References
A.S. Ahmed, S.M. Muhamed, M.L. Singla, S. Tabassum, A.H. Naqvi, A. Azam, Band gap narrowing and fluorescence properties of nickel doped SnO2 nanoparticles. J. Lumin. 131, 1–6 (2011)
S.Y. Choi, M.H. Kim, Y. Kwon, Effects of Cr2O3 modification on the performance of SnO2 electrodes in DSSCs. Phys. Chem. Chem. Phys. 14, 3576 (2012)
S. Kumar, S. Bhunia, A.K. Ojha, Effect of calcination temperature on phase transformation, structural and optical properties of sol–gel derived ZrO2 nanostructures. Physica E 66, 74 (2015)
M.C. Mathpal, P. Kumar, R. Balasubramaniyan, J.S. Chung, A.K. Tripathi, M.K. Singh, M.M. Ahmad, S.N. Pandey, A. Agarwal, Facile deposition and plasmonic resonance of Ag–Au nanoparticles in titania thin film. New J. Chem. 39(8), 6522–6530 (2015)
V. Agrahari, M.C. Mathpal, M. Kumar, A. Agarwal, Investigations of optoelectronic properties in DMS SnO2 nanoparticles. J. Alloy. Compd. 622, 48 (2015)
K.L. Chopra, S. Major, D.K. Pandya, Transparent conductors-A status review. Thin Solid Films 102, 1 (1983)
Z. Ying, Q. Wan, Z.T. Song, S.L. Feng, SnO2 nanowhiskers and their ethanol sensing characteristics. Nanotechnology 15, 1682 (2004)
A. Aoki, H. Sasakura, Tin oxide thin film transistors. J. Appl. Phys. 9, 582 (1970)
L.P. Singh, N.M. Luwang, S.K. Srivastava, Luminescence and photocatalytic studies of Sm3+ ion doped SnO2 nanoparticles. New J. Chem. 38, 115–121 (2014)
K. Lodewijks, N. Maccaferri, T. Pakizeh, R.K. Dumas, I. Zubritskaya, J. Akerman, P. Vavassori, A. Dmitriev, Magnetoplasmonic design rules for active magneto-optics. Nano Lett. 14(12), 7207–7214 (2014)
R.K. Selvan, I. Perelshtein, N. Perkas, A. Gedanken, Synthesis of hexagonal shaped SnO2 nanocrystals and SnO2 nanocomposites for electrochemical redox supercapacitors. J. Phys. Chem. C 2008, 112 (1825)
N. Maccaferri, K.E. Gregorczyk, V.A. Thales, G.D. Oliveira, M. Kataja, S.V. Dijken, Z. Pirzadeh, A. Dmitriev, J. Akerman, M. Knez, P. Vavassori, Ultrasensitive and label-free molecular-level detection enabled by light phase control in magnetoplasmonic nanoantennas. Nat. Commun. (2015). doi:10.1038/ncomms7150
M. Johnson, Introduction to magnetoelectronics (AP Academic press, Waltham, 2004), pp. 1–65
J. Huang, A. Lu, B. Zhao, Q. Wan, Branched growth of degenerately Sb-doped SnO2 nanowires. Appl. Phys. Lett. 91, 073102 (2007)
C. Drake, S. Seal, Band gap energy modifications observed in trivalent in substituted nanocrystalline SnO2. Appl. Phys. Lett. 90, 233117 (2007)
S.S. Pan, G.H. Li, L.B. Wang, Y.D. Shen, Y. Wang et al., Atomic nitrogen doping and p-type conduction in SnO2. Appl. Phys. Lett. 95, 222112 (2009)
C.X. Hu, Y.S. Wu, H.Y. Wei, Y.C. Shi, L.L. Wu, A comparative study of the effects of Cu, Rh doping on the microstructure, morphological and optical properties of tin dioxide nanocrystallines. J. Mater. Sci. 40, 6301–6306 (2005)
Y.S. Kim, S.G. Ansari, Z.A. Ansari, R. Wahab, H.S. Shin, A simple method to deposit palladium doped SnO2 thin films using plasma enhanced chemical vapor deposition technique. Rev. Sci. Instrum. 81, 113903 (2010)
O. Mounkachi, E. Salmani, H.E. Moussaoui, M. Hamedoun, E.H. Zahraouy, A. Benyoussef, Ab-initio calculation of half-metal ferrimagnetic Sn0.9Mn0.05Co0.05O2. J. Supercond. Novel Magn. 25, 2069–2074 (2012)
A.C. Yanes, M.J. Ramos, J.D. Castillo, J.J. Velázquez, V.D. Rodríguez, Size-dependent luminescence of Sm3+ doped SnO2 nano-particles dispersed in sol-gel silica glass. Appl Phys B 101, 849–854 (2010)
B. Sathyaseelan, K. Senthilnathan, T. Alagesan, R. Jayavel, K. Sivakumar, A study on structural and optical properties of Mn and Co doped SnO2 nanocrystallites. Mater. Chem. Phys. 2010, 124 (1046)
D. Wang, J. Jin, D. Xia, Q. Ye, J. Long, The effect of oxygen vacancies concentration to the gas-sensing properties of tin dioxide-doped Sm. Sens. Actuators B 66, 260–262 (2000)
A.K. Tripathi, M.C. Mathpal, P. Kumar, M.K. Singh, S.K. Mishra, R.K. Srivastava, J.S. Chung, G. Verma, M.M. Ahmad, A. Agarwal, Synthesis based structural and optical behavior of anatase TiO2 nanoparticles. Mater. Sci. Semicond. Process. 23, 136–143 (2014)
A.K. Tripathi, M.K. Singh, M.C. Mathpal, S.K. Mishra, A. Agarwal, Study of structural transformation in TiO2 nanoparticles and its optical properties. J. Alloys Compd. 549, 114–120 (2013)
A. Dieguez, A.R. Rodriguez, A. Vila, J.R. Morante, The complete Raman spectrum of nanometric SnO2 particles. J. Appl. Phys. 90, 1550 (2001)
H.C. Choi, Y.M. Jung, S.B. Kim, Size effects in the Raman spectra of TiO2 nanoparticles. Vib. Spectrosc. 37(1), 33–38 (2005)
A.A. Roselin, N. Anandhan, G. Ravi, M. Mummoorthi, T. Marimuthu, Growth and characterization of Sm2O3 thin films by spin coating technique. Int. J. ChemTech Res. 6(13), 5315–5320 (2014)
G.G. Xi, J. Ye, Ultrathin SnO2 Nanorods: template and surfactant-free solution phase synthesis, growth mechanism, optical, gas-sensing, and surface adsorption properties. Inorg. Chem. 49, 2302–2309 (2010)
M.C. Mathpal, A.K. Tripathi, M.K. Singh, S.P. Gairola, S.N. Pandey, A. Agarwal, Effect of annealing temperature on Raman spectra of TiO2 nanoparticles. Chem. Phys. Lett. 555, 182–186 (2013)
X. Feng, J. Ma, F. Yang, F. Ji, F. Zong, C. Luan, H. Ma, Structural and UV photoluminescence properties of single crystalline SnO2 films grown on α - Al2O3 (0001) by MOCVD. Solid State Commun. 144, 269 (2007)
C. Wang, M. Ge, J.Z. Jiang, Magnetic behavior of SnO2 nanosheets at room temperature. Appl. Phys. Lett. 97, 042510 (2010)
G.A. Alanko, A. Thurber, C.B. Hanna, A. Punnoose, Size, surface structure, and doping effects on ferromagnetism in SnO2. J. Appl. Phys. 111, 07C321 (2012)
P. Wu, B. Zhou, W. Zhou, Room-temperature ferromagnetism in epitaxial Mg-doped SnO2 thin films. Appl. Phys. Lett. 100, 182405 (2012)
B. Huang, D. Zhu, X. Ma, Great influence of the oxygen vacancies on the ferromagnetism in the Co-doped ZnO films. Appl. Surf. Sci. 253, 6892–6895 (2007)
S. Sambasivam, D.P. Joseph, J.H. Jeong, B.C. Choi, K.T. Lim, S.S. Kim, T.K. Song, Antiferromagnetic interactions in Er-doped SnO2 DMS nanoparticles. J. Nanopart. Res. 13, 4623–4630 (2011)
S. Kumar, A.K. Ojha, Oxygen vacancy induced photoluminescence properties and enhanced photocatalytic activity of ferromagnetic ZrO2 nanostructures on methylene blue dye under ultra-violet radiation. J. Alloys Compd. 644, 654–662 (2015)
V. Agrahari, A.K. Tripathi, M.C. Mathpal, A.C. Pandey, S.K. Mishra, R.K. Shukla, A. Agarwal, Effect of Mn doping on structural, optical and magnetic properties of SnO2 nanoparticles. J. Mater. Sci. Mater. Electron. (2015). doi:10.1007/s10854-015-3620-0
Acknowledgments
One of the authors is thankful to Prof. Avinash C. Pandey, Nano Application Centre, University of Allahabad, and Prof. Ramesh Chandra, IIT Roorkee for getting characterizations of samples and their valuable discussion for the research work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Agrahari, V., Mathpal, M.C., Kumar, S. et al. Low field room temperature magnetism and band gap modifications in Sm doped SnO2 . J Mater Sci: Mater Electron 27, 3053–3064 (2016). https://doi.org/10.1007/s10854-015-4129-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-015-4129-2