
Abstract
To evaluate the effect of chemical structure on flame-retardant mechanisms, two types of flame retardants (FRs) containing both phosphorus and nitrogen groups such as N-(2-methoxyethyl)-N-methyl-P,P-diphenylphosphinic amide (PNOFR) and 2-(dimethylamino)ethyl diphenylphosphinate (PONFR) were synthesized and applied to polyvinyl chloride (PVC). PNOFR and PONFR exhibited the typical flame-retardant mechanism of FRs containing phosphorus and nitrogen compounds. It was observed that PNOFR and PONFR have different main flame-retardant mechanisms depending on the element combined with phosphorus in their chemical structures. In the condensed phase, PNOFR induced a denser crosslinked char layer with a higher phosphorus content compared with PONFR. By contrast, in the gas phase, PONFR released more PO2∙ and PO∙ radicals compared with PNOFR. Therefore, while PNOFR and PONFR have flame-retardant properties through both mechanisms, the dominant mechanisms for PNOFR and PONFR are different. This study presents an effective strategy for designing attractive chemical structures to prepare FRs for applications in various polymers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Flame retardants (FRs) are materials that are introduced into combustible materials, such as polymers, to inhibit the spreading of fire and provide additional escape time [1,2,3]. Halogenated FRs have been widely applied in various industries because of their high flame-retardant efficiency and low cost. However, despite these advantages, halogenated FRs have recently been banned in many countries because of environmental problems; their materials emit toxic and corrosive gases during combustion [4,5,6]. Accordingly, halogen-free FRs containing aluminum, manganese [7,8,9], silicon [10, 11], and organo-phosphorus compounds [12, 13] have attracted considerable attention. Among these halogen-free FRs, because FRs containing organophosphorus compounds show better compatibility with polymers than FRs containing other inorganic compounds, many studies on FRs containing organophosphorus compounds have been conducted [14,15,16,17]. Nevertheless, it is still not sufficient to endow polymers with superior flame retardancy by introducing a single flame-retardant element.
FRs containing phosphorus and nitrogen compounds are widely known to be effective in imparting flame retardancy to polymers via the synergistic effect between phosphorus and nitrogen [18]. For phosphorus in this system, the flame-retardant mechanism works in both the condensed and gas phases [19]. In the gas phase, the FRs are decomposed at a relatively low temperature during combustion and generate active radicals (PO∙, PO2∙, and HPO∙), which can scavenge flammable components, such as H∙ and ∙OH, in air [5, 19, 20]. In the condensed phase, phosphoric acid or polyphosphoric acid, which is produced by the decomposition of FRs during combustion, acts as catalysts and assist in the formation of a carbonaceous layer on the surface by dehydrating the polymer. This layer restricts the heat transfer and contact of oxygen and combustible gases to the polymers, shielding the combustion [19,20,21]. The flame-retardant mechanism mainly works in the gas phase for nitrogen in the system. The FRs are decomposed into non-toxic and non-combustible gases, such as nitrogen oxide and ammonia, and dilute the concentration of oxygen in the atmosphere to extinguish the flame [22, 23]. Consequently, because the flame-retardant mechanisms of both phosphorus and nitrogen can simultaneously contribute, FRs containing phosphorus and nitrogen compounds could maximize the flame retardancy compared to each single flame retardant system. Accordingly, some studies on the synthesis and synergistic flame retardant effect of FRs containing phosphorus and nitrogen compounds have been reported, and the flame retardant effect was shown by the formation of char layer in the condensed phase [24,25,26,27]. Also, it has been studied that the inclusion of oxygen and phosphorus in the chemical structure reduced the effective heat of combustion and a gas phase mechanism occurred [28, 29]. However, there is little research on the flame retardant effect by the accurate structure for the flame retardant mechanism. Therefore, we
Herein, to systematically investigate the flame-retardant effect depending on the chemical bonds in the structure of FRs, two types of FRs containing phosphorus and nitrogen compounds with the same chemical formula were designed and synthesized. One FR has a structure in which phosphorus and nitrogen are directly combined, and the other FR has a structure in which phosphorus and nitrogen are separate. The intention is to check whether the phosphorus-nitrogen bond influences the flame retardant effect and mechanism. In addition, because these FRs have identical phosphorus and nitrogen contents, the influence of phosphorus and nitrogen content on the flame-retardant mechanism is excluded. Thus, the flame-retardant effect according to the difference in the chemical bonding of each FR was closely investigated. For comparison, poly(vinyl chloride) (PVC), one of the common thermoplastics, was used as a polymer matrix. It is widely known that PVC has mainly a gas-phase pyrolysis mechanism, which emits HCl and forms conjugated network [30,31,32,33]. Further, in condensed phase, char layer is generated by cross-linking reaction of the conjugated polymer chains during this consecutive pyrolysis process [33, 34]. Therefore, we determined that PVC is a suitable matrix for analyzing the flame retardancy and flame retardant mechanism of two FRs in both gas and condensed phase. The flame retardancy and combustion behavior of the two FRs were evaluated using limiting oxygen index (LOI) and vertical burning test (UL-94). In addition, the flame-retardant mechanism according to the chemical structure of the two FRs was investigated using thermogravimetric analysis-infrared spectrometry (TG-IR), pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS), field emission scanning electron microscope (FE-SEM), and Raman spectroscopy (RAMAN).
Materials and methods
Materials
Diphenyl phosphinic chloride and N-(2-methoxyethyl)methylamine were purchased from TCI. 2-Dimethyl aminoethanol and polyvinylchloride (PVC) (Mw ~ 43,000 g mol−1, Mn ~ 22,000 g mol−1, inherent viscosity 0.5 Dl g−1) were purchased from Sigma-Aldrich. Tetrahydrofuran (THF), methylene chloride (MC), magnesium sulfate (MgSO4), and sodium hydroxide (NaOH) were purchased from Daejung Chemical & Metals. Triethylamine (TEA) was purchased from Junsei Chemical Co. Ltd. All other reagents and solvents were used as received, without further purification.
Synthesis of N-(2-methoxyethyl)-N-methyl-P,P-diphenylphosphinic amide (PNOFR)
N-(2-methoxyethyl)methylamine (0.1 mol) and TEA (0.1 mol) were dissolved in THF in a round-bottom flask with magnetic stirring. The mixture was cooled in an ice bath, and diphenyl phosphinic chloride (0.1 mol) solution dissolved in THF was added dropwise into the flask for approximately 1 h. Further, the reaction mixture was stirred for 24 h at 25 °C. After the reaction was complete, the reaction mixture was filtered to remove triethylamine hydrochloride, and the solution was concentrated on a rotary evaporator. The liquid was diluted with MC, extracted 3–4 times with 1 M NaOH solution and extracted 3–4 times with distilled water. The MC layer was dried with anhydrous MgSO4, and MC was removed using a rotary evaporator to obtain pure PNOFR (yield: 62%). 1H NMR (400 Hz, CDCl3, ppm, δ): 7.8–7.4 (m, 10H, P–Ph), 3.5 (t, 2H, –CH2–O–), 3.2 (d, 3H, –O–CH3), 3.1 (m, 2H, –N–CH2–), 2.7 (t, 3H, –N–CH3).
Synthesis of 2-(dimethylamino)ethyl diphenylphosphinate (PONFR)
2-Dimethyl aminoethanol was added instead of N-(2-methoxyethyl)methylamine, and other experiments and purification procedures were the same as those for PNOFR (yield: 55%). 1H NMR (400 Hz, CDCl3, ppm, δ): 7.8–7.4 (m, 10H, –P–Ph), 4.1 (m, 2H, –O–CH2–), 2.6 (t, 2H, –CH2–N–), 2.2 (s, 6H, –N–CH3).
Preparation of PVC films
The PVC films were prepared using a solution casting method as follows: PVC (20 g) and different amounts of FRs (PNOFR and PONFR) were dissolved in 100 mL of THF to prepare PVC films containing 10 and 20 wt% of each FR (Table 1). After mixing for 1 h, the transparent solution was placed in a glass mold and dried overnight at 25 °C. Subsequently, the sample was dried at 40 °C in a vacuum oven for two days to remove the residual solvent. The PVC films containing 10 and 20 wt% FRs were labeled PVC-PNOFR# and PVC-PONFR#, respectively, where # is the amount of FRs.
Characterization
1H nuclear magnetic resonance (1H NMR) spectroscopy was performed with a Bruker AVANCE 400 MHz NMR spectrometer using CDCl3 as the solvent. 31P nuclear magnetic resonance (31P NMR) spectra were recorded using a Bruker AVANCE-500 MHz NMR spectrometer. LOI was determined using a FESTEC FT-LOI-404 LOI tester, according to the ASTM D2863 standard. The dimension of the sample was 120 mm × 10 mm × 1.3 mm. UL-94 test was performed according to ASTM D3801 with a sample of dimension of 125 mm × 12 mm × 1.0 mm. Fourier transform infrared (FTIR) spectra were obtained using a Bruker Alpha-Platinum attenuated total reflectance (ATR)-FTIR spectrometer over the range 4000–400 cm−1. The thermal properties of the flame retardants were analyzed by thermogravimetric analysis (TGA) from 25 to 700 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere using a Perkin Elmer TGA 4000 instrument. The change in chemical structure with temperature was analyzed using TGA and FTIR. After heating to the required temperature by TGA in air, the chemical structures of the samples were determined by FTIR. For the samples burned at 500 °C for 10 min, the morphology of the residual char was investigated using a JEOL JSM-7601F PLUS FE-SEM. The surfaces of samples were coated with a platinum layer. SEM–energy dispersive X-ray spectroscopy (SEM–EDS) was performed to confirm the phosphorus content of the residual char. RAMAN measurements were performed using a Thermo Fisher DXR2xi instrument with a 532 nm laser line at 25 °C. Py-GC/MS analysis was performed using a single-shot Agilent system. The temperature of the GC/MS interface was 300 °C and the cracker temperature was 400 °C. The gas chromatogram was obtained using a capillary column (DB5, 30 m × 0.25 mm × 0.25 mm) with high-purity helium as the carrier gas (1.0 mL min−1).
Results and discussion
Characterization of PNOFR and PONFR
To examine the flame-retardant mechanism depending on the chemical structure of FRs containing phosphorus and nitrogen elements, we designed and synthesized two types of FRs (PNOFR and PONFR) with different chemical bonding and the same chemical formula (Scheme 1). The chemical structure of each FR was investigated using FTIR, 1H NMR, and 31P NMR spectroscopy (Fig. 1). The FTIR spectra of the PNOFR and PONFR are shown in Fig. 1a. For both PNOFR and PONFR, the common specific vibrations assigned to the C–C stretching of phenyl groups (1591 cm−1), P–Ph stretching (1441 cm−1), C–N stretching (1368 cm−1), P=O stretching (1216 cm−1), C=C stretching of the aromatic ring (825 cm−1), and P–C (696 cm−1) were observed. Meanwhile, in the case of PNOFR, peaks at 1021 and 925 cm−1 ascribed to the vibration of P–N stretching were observed, indicating a successful condensation reaction between diphenylphosphinic chloride and N-(2-methoxyethyl)methylamine. In the case of PONFR, the vibration attributed to P–O stretching appeared at 1130 cm−1, indicating that a reaction occurred between diphenylphosphinic chloride and 2-dimethyl aminoethanol. This demonstrated that two phosphorus/nitrogen FRs were successfully synthesized. Furthermore, the 1H NMR and 31P NMR spectra clearly show the chemical structures of the two FRs, as shown in Fig. 1b, c. The 31P NMR spectra of PNOFR and PONFR show a chemical shift at 29.2 and 29.9 ppm, respectively, and no peak corresponded to diphenylphosphinic chloride. In addition, PNOFR and PONFR appeared as single peaks in the 31P NMR spectra, indicating that PNOFR and PONFR were successfully synthesized with high purities.

Different types of phosphorus/nitrogen flame retardant

a FTIR spectra of PNOFR and PONFR, b 1H NMR spectra of PNOFR and PONFR, and c 31P NMR spectra of diphenyl phosphinic chloride, PNOFR, and PONFR
Flame retardancy of PVC-PNOFR and PVC-PONFR films
To confirm the flame-retardant effect according to the chemical structure of the FRs, a LOI test and UL-94 test were carried out for the PVC-PNOFR and PVC-PONFR films (Fig. 2). In order that measurement the flame retardancy of the flame retardant, poly(vinyl chloride) (PVC) which is one of the most common thermoplastic polymers used in a wide range of applications such as pipes, wire, and cable was chosen as the polymer matrix. PVC films were prepared by introducing PNOFR and PONFR into PVC through the solution-casting method. The PVC films were labeled PVC-PNOFR# and PVC-PONFR#, where # is the weight percentage of each phosphorus/nitrogen FR. The UL-94 test results showed that neat PVC was completely burned with severe melt-dripping behavior at the first ignition and failed to pass the UL-94 test. By contrast, when PNOFR and PONFR were introduced into PVC, the PVC films exhibited self-extinguishing properties within 10 s at the first and second ignition, regardless of the type and amount of FRs, resulting in a V-0 rating. In addition, both the PVC-PNOFR and PVC-PONFR films were extinguished with no melt-dripping behavior after ignition, indicating that PNOFR and PONFR impart flame retardancy and anti-dripping properties to PVC. The LOI test results also indicated a similar flame retardancy for PNOFR and PONFR. The LOI value of neat PVC was 24.2%, whereas those of PVC-PNOFR10 and PVC-PONFR10 were 26.7 and 26.6%, respectively, indicating that PNOFR and PONFR induce flame retardancy in PVC. This result was also obtained even when the amounts of PNOFR and PONFR were increased to 20 wt%: PVC-PONFR20 (27.5%) ≈ PVC-PNOFR20 (27.0%). The LOI results showed that while flame retardancy improved with increasing FR content, no significant difference was observed in the flame-retardant effect depending on the chemical structure. It has been previously reported that the P–N bond in FRs improves the flame-retardant effect. However, contrary to our expectations, both PNOFR and PONFR show similar flame retardant effects on PVC even there is no P–N bond in PONFR.

LOI values and UL-94 test rating of neat PVC, PVC-PNOFR, and PVC-PONFR films
Therefore, the chemical structures of neat PVC, PVC-PNOFR20 and PVC-PONFR20 during the thermal degradation process were investigated by TGA and FTIR to confirm the flame-retardant role of PNOFR and PONFR in PVC (Fig. 3). For the experiments, neat PVC, PVC-PNOFR20 and PVC-PONFR20 were heated to the required temperature using TGA under an air atmosphere, and then, the chemical structure during the thermo-oxidative degradation was observed using FTIR. For neat PVC film, the characteristic peaks of PVC assigned to C–Cl stretching (687 cm−1), –CH– bending (1248 cm−1), –CH2 bending (1428 cm−1) and –CH2 asymmetric or symmetric stretching (2800–3000 cm−1) were observed (Fig. 3a) [35, 36]. As the temperature increased, the intensity of characteristic peaks gradually decreased and disappeared completely at 300 °C. In addition, new peak attributed to C=C stretching (1579 cm−1) was observed at 300 °C and intensity of the peak increased with the raise of temperature, indicating that the structure of PVC was almost decomposed by dehydrochlorination and aliphatic structure and/or aromatic rings of C=C were formed during thermal degradation process [37]. PVC-PNOFR20 and PVC-PONFR20 show similar decomposition behavior to neat PVC, with slight differences at temperatures above 300 °C. As shown in Fig. 3b, the peaks at 925 and 1021 cm−1 are corresponding to P–N stretching of PNOFR and disappeared at 400 °C. From the Fig. 3c, the P=O stretching (1223 cm−1), P–O stretching (1130 cm−1) and C=C stretching of the aromatic ring (825 cm−1) of PONFR became weak and disappeared completely at 250 °C. As the temperatures increased above 250 °C, several common peaks for PVC-PNOFR20 and PVC-PONFR20 were observed. The P=O stretching (1178 and 1127 cm−1) appeared at 250 °C. In addition, P–O–Ar stretching (988 cm−1) and P–O–P symmetric/asymmetric stretching (867 cm−1) were observed at 450 °C, indicating the formation of phosphoric acid or polyphosphoric acid, which promote the degradation of polymer matrix and stable char formation of polyaromatic structures [38, 39]. In contrast to neat PVC, C–Cl stretching, –CH– bending, –CH2 bending and –CH2 asymmetric or symmetric stretching, which corresponding to the characteristic peaks of PVC, disappeared at 400 and 450 °C. Moreover, the C=C stretching vibration peak was observed at 200 °C and 250 °C in PVC-PNOFR20 and PVC-PONFR20, respectively. Consequently, as PNOFR and PONFR degraded into phosphoric acid or polyphosphoric acid, promote the decomposition of the polymer matrix and block contact with heat and oxygen by formation of the stable char with polyaromatic structures to inhibit PVC dehydrochlorination and delay thermal decomposition. Nevertheless, there was an obvious difference between PNOFR and PONFR at the temperature which C=C peak appeared; PNOFR observed 200 °C and PONFR observed 250 °C. It was estimated that the temperature of stable char formation for the two flame retardants were different, which was considered as a difference in the decomposition behavior according to the chemical structure of the flame retardant. Hence, to confirm the cause of these results, the condensed-phase and gas-phase FR mechanisms of each FR were investigated in detail. Then, we further focused on the flame-retardant mechanism according to the chemical structure of the FRs [40].

FTIR spectra of the char layer of a neat PVC, b PVC-PNOFR20 and b PVC-PONFR20 at different temperature
Condensed-phase flame retardant mechanism of PNOFR and PONFR
First, to investigate how PNOFR and PONFR affect the formation of the char layer in the condensed phase during the combustion process of a polymer matrix, the morphology of the char layer of the PVC films was examined using SEM (Fig. 4). For the preparation of the samples, the neat PVC, PVC-PNOFR20, and PVC-PONFR20 were heated at 500 °C for 10 min in a furnace. As shown in Fig. 4, the char of neat PVC exhibited a fractured structure with many holes, which could easily release HCl and flammable volatile gas. Thus, this structure cannot prevent heat and mass transfer from/to the surface during combustion, which leads to flammability. Meanwhile, there was a difference in the char morphology based on the type of flame retardant used in the PVC. In the case of PVC-PNOFR20, it formed a compact and dense char structure without holes, indicating that it inhibited heat transfer. In other words, PNOFR imparted superior flame retardancy to PVC in its condensed phase. By contrast, although PVC-PONFR20 formed a harder and thicker char structure than neat PVC, it had some pores with small sizes, suggesting that PONFR cannot completely restrain the transfer of heat and oxygen. Thus, PNOFR showed a more effective condensed-phase flame-retardant mechanism by inducing a more stable char layer compared to PONFR, which is also supported by Raman spectroscopy. Raman spectroscopy was performed to characterize the degree of graphitization of the char residues.

SEM images and EDS mapping spectra of the residual char layer of a neat PVC, b PVC-PNOFR20, and c PVC-PONFR20
As shown in Fig. 5, all samples had two broad absorption peaks: a peak at approximately 1373 cm−1 (D band) assigned to disordered graphite and a peak at approximately 1597 cm−1 assigned to organized graphite [41,42,43,44]. The intensity ratio of the D and G bands (ID/IG) can be used to estimate the graphitization degree of char; furthermore, a lower value of ID/IG indicates a higher degree of graphitization [45]. The ID/IG values of neat PVC, PVC-PNOFR20, and PVC-PONFR20 were 2.00, 1.75, and 1.91, respectively. Among the PVC films, PVC-PNOFR20 showed the lowest ID/IG value, demonstrating that PNOFR facilitates the formation of a more graphitized carbon structure compared to PONFR. Considering that the graphitized carbon structure effectively prevents heat and oxygen during combustion [46, 47], PNOFR is more favorable for a more stable char formation in the condensed phase compared to PONFR. These results were in accordance with the SEM results. Additionally, from the SEM–EDS spectra in Fig. 4, the phosphorus content of the char residue of PVC-PNOFR20 was higher than that of PVC-PONFR20. Despite having the same chemical formula, the elemental composition of the char residue differed according to the chemical structure. While PNOFR contributed to char formation with a high phosphorus content, PONFR decomposed to PO∙ and PO2∙ radicals. Therefore, the phosphorus content in the char residue of PVC-PONFR20 was lower than that of PVC-PNOFR20. This is further discussed in gas-phase flame retardant mechanism section that follow. Consequently, PNOFR has a flame-retardant effect through the condensed phase in the two types of flame-retardant mechanisms. That is, the condensed-phase flame-retardant mechanism of PNOFR is superior to that of PONFR.

Raman spectra of the residual char layer of a neat PVC, b PVC-PNOFR20, and c PVC-PONFR20
Furthermore, to supplement the flame retardant mechanism of PNOFR and PONFR in the condensed phase, the thermo-oxidative degradation behaviors of PNOFR and PONFR were analyzed using TG-IR. The heat treatment temperature was set in consideration of the thermal degradation temperatures of the PNOFR and PONFR (Fig. S1 and Fig. 6). As shown in Fig. 6, the new peaks at 1645 and 1643 cm−1 assigned to the C=C stretching vibration of aromatic rings were observed at 150 °C for PNOFR and PONFR, respectively. In addition, the peak intensity at 2750–3100 cm−1 attributed to the –CH3 and –CH2 stretching vibrations gradually decreased with increasing the temperature and disappeared at 350 °C. This indicates that thermo-oxidative degradation process of PNOFR and PONFR begins and forms the residual char with aromatic and polyaromatic structures in the temperature range from 150 to 350 °C. Therefore, analysis of the thermo-oxidative degradation behavior of PNOFR and PONFR focused on the temperature range from 150 to 350 °C. From the FTIR spectra of PNOFR in Fig. 6a, the P–N stretching (1021 cm−1) gradually decreased and disappeared completely at 400 °C, and another P–N peak at 925 cm−1 disappeared at 150 °C. Furthermore, the P=O stretching (1216 cm−1) was shifted to 1172 and 1231 cm−1 at 150 and 350 °C, respectively, implying the formation of phosphoric acid via thermal degradation [48,49,50]. New peaks attributed to P–O–P symmetric and asymmetric stretching (983 and 924 cm−1) were observed at 150 °C. In addition, other new peaks attributed to P–O–P stretching (865 cm−1) and P–O–Ar stretching (949 cm−1) appeared at above 300 °C. These results indicate that the thermo-oxidative degradation of PNOFR induces the cleavage of the P–N bond, resulting in the formation of phosphoric acid and polyphosphoric acid, which promotes the dehydration condensation reaction to form a crosslinked-aromatic structure [51,52,53]. As shown in Fig. 6b, PONFR exhibited a thermo-oxidative degradation behavior similar to that of PNOFR. The P–O stretching (1130 cm−1) disappeared above 200 °C, and the peak assigned to P=O stretching (1216 cm−1) shifted to 1181 cm−1 above 200 °C. Moreover, the peaks contributed to P–O–P symmetric/asymmetric stretching (858 cm−1) and P–O–Ar stretching (953 cm−1) were newly observed at 300 °C. Thus, the PNOFR and PONFR thermally degraded and formed phosphoric acid and polyphosphoric acid, which facilitated the formation of a stable char layer. This indicates that PNOFR and PONFR induce the formation of a stable and rigid crosslinked char layer, leading to the effective prevention of heat and oxygen transfer during combustion, which is the same as the TG-IR results of PNOFR and PONFR in PVC analyzed previously.

FTIR spectra of the condensed phase of a PNOFR and b PONFR at different temperature
Gas-phase flame retardant mechanism of PNOFR and PONFR
To confirm the retardant mechanism of PNOFR and PONFR in the gas phase, pyrolysis-GC/MS was used [54,55,56]. The cracker temperature was selected as 400 °C, at which PNOFR and PONFR were completely decomposed (Fig. S1). The pyrolysis-GC/MS chromatograms and the main thermal decomposition products are summarized in Fig. 7 and Table 2, respectively. The cracking of both PNOFR and PONFR was mainly accompanied by the formation of small molecules and diphenyl phosphine derivatives. The small molecules were further decomposed into non-combustible gases, such as CO2 and N2, which might dilute the concentrations of oxygen and volatile gases in the air. Notably, some differences in the formation of diphenyl phosphine derivatives between PNOFR and PONFR are closely related to the differences in the chemical structure of FRs [57, 58]. In the pyrolysis of PNOFR, the cleavage of the P–N bond leads to the formation of diphenyl phosphine derivatives containing oxygen or radicals (m/z = 201, 202, and 217). Furthermore, the condensation reaction between these diphenyl phosphine derivatives occurred, resulting in the formation of polyphosphoric acid with P–O–P bonds. It is widely known that P–O–P bonds are involved in the carbonization process as crosslinkers that link different carbon-based materials. These crosslinked carbon structures contribute to the formation of a dense char layer, meaning that the pyrolysis products of PNOFR participate in char formation through the condensed-phase flame retardant mechanism of FRs containing phosphorus. By contrast, while PONFR also decomposed into diphenyl phosphine derivatives containing oxygen or radicals, some diphenyl phosphine derivatives were further cracked to benzene and organophosphorus radicals. Subsequently, by the elimination of benzene, the organophosphorus radicals generate PO2∙ and PO∙ radicals rather than polyphosphoric acid, which could serve as a scavenger for H∙ and ∙OH in the air. As shown in Fig. 7, the peak attributed to the PO∙ radical was observed at the 10 and 7 peak positions in the PONFR and PNOFR chromatograms, respectively [59,60,61]. Compared with the PNOFR chromatogram, the peak intensity of PONFR (10 peak position) was relatively larger than that of PNOFR (7 peak position). In addition, while the peak contributing to the PO2∙ radical was observed at 4 peak position in the PONFR chromatogram, no corresponding peak was observed in the PNOFR chromatogram. This means that the flame-retardant effect of PONFR is superior in the gas phase compared with that of PNOFR. In other words, PONFR predominantly form gaseous pyrolysis products, such as PO2∙ and PO∙ radicals, rather than condensed pyrolysis products, such as phosphoric acid, during combustion. This supports the fact that compared to PNOFR, PONFR has a lower ID/IG value in the Raman results and a lower phosphorus content in the char in the SEM–EDS results.

Pyrolysis-GC/MS chromatograms of a PNOFR and b PONFR at the pyrolysis temperature of 400 °C
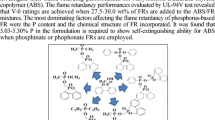
Based on the above analysis, the flame-retardant mechanism according to the decomposition behavior of PNOFR and PONFR is explained and presented in Fig. 8. PNOFR and PONFR showed typical flame-retardant mechanisms of FRs containing phosphorus and nitrogen compounds in both condensed and gas phases. In general, PNOFR and PONFR generated inert gases from the nitrogen source, regardless of the chemical structure. Meanwhile, it was revealed that PNOFR and PONFR have different main flame-retardant mechanisms (condensed phase and gas phase) depending on the element combined with phosphorus. In the gas phase, while both PNOFR and PONFR showed gaseous pyrolysis products, such as PO∙ radicals, which could scavenge the active radicals in the air, the relative amounts of products were much higher in PONFR than in PNOFR. In addition, PONFR generates radicals containing phosphorus, which are not found in the pyrolysis product of PNOFR. Accordingly, for PONFR, the degree of polyphosphoric acid formation decreased in the condensed phase. On the other hand, for PNOFR, although few radicals containing phosphorus were observed in the gas phase, the cleavage of the P–N bond mainly led to the formation of polyphosphoric acid, which promoted the crosslinked char layer of the polymer matrix in the condensed phase.

Decomposition behavior of a PNOFR and b PONFR in the condensed and gas phases
Because the phosphorus and nitrogen contents of PNOFR and PONFR are the same, these results indicate that the difference in the flame retardant mechanisms only contributes to the chemical structure. Although the two FRs have different flame-retardant mechanisms, they showed similar flame-retardant effects in the LOI and UL-94 test results. This means that the two flame-retardant mechanisms are complementary. For example, the low flame-retardant effect of PONFR, forming less dense char in the condensed phase, is further compensated by the generation of a large amount of active radicals in the gas phase. By contrast, PNOFR shows a flame-retardant effect via the formation of dense and stable char despite the release of a small amount of active radicals. Therefore, if we design a chemical structure in which both flame-retardant mechanisms can act effectively, the flame-retardant effect would be significantly increased.
Conclusion
In summary, two FRs (PNOFR and PONFR) containing phosphorus and nitrogen with the same chemical formula and different chemical structures were synthesized to demonstrate the flame-retardant mechanism depending on the chemical structure. PNOFR and PONFR are structures having nitrogen and oxygen that combined with phosphorus, respectively. The results of the LOI and UL-94 tests for the two FRs were similar. Therefore, the flame-retardant mechanisms of PNOFR and PONFR were systematically investigated in terms of their condensed and gas phases. TG-IR analysis showed that PNOFR and PONFR formed a phosphorus-rich and dense crosslinked char layer. In addition, the residual char of PNOFR showed a higher phosphorus content and graphitization degree compared to that of PONFR, indicating that PNOFR effectively prevents heat and oxygen transfer during combustion in the condensed phase. Pyrolysis-GC/MS results revealed that PNOFR mainly formed polyphosphoric acid, which facilitated the stable char formation of the polymer matrix. By contrast, PONFR primarily formed PO2∙ and PO∙ radicals, which could scavenge H∙ and ∙OH in air rather than polyphosphoric acid. Consequently, although PNOFR and PONFR showed a typical flame retardant mechanism in both the condensed and gas phases, dominant condensed-phase and gas-phase flame retardant mechanisms have been observed, respectively. Furthermore, this study provides the direction of structural design according to condensed or gas-phase flame-retardant mechanisms required for application to various polymer matrices.
Data availability
Not Applicable.
Code availability
Not Applicable.
References
He W, Song P, Yu B, Fang Z, Wang H (2020) Prog Mater Sci 114:100687
Laoutid F, Bonnaud L, Alexandre M, Lopez-Cuesta J-M, Dubois P (2009) Mater Sci Eng R Rep 63:100–125
Lim K-S, Bee S-T, Sin LT et al (2016) Compos B Eng 84:155–174
Levchik SV, Weil ED (2006) J Fire Sci 24:345–364
Zhang W, He X, Song T, Jiao Q, Yang R (2014) Polym Degrad Stab 109:209–217
Li L, Wang H, Hua F et al (2021) Macromol Res 29:625–635
Wang X, Kalali EN, Wang D-Y (2016) Nano Adv 1:1–16
Cheng JJ, Shi BB, Zhou FB, Chen XY (2014) J Appl Polym Sci 131:40253
Guo X, Wang H, Ma D, He J, Lei Z (2018) J Appl Polym Sci 135:46410
Hsiue GH, Wang WJ, ChangSupSup FC (1999) J Appl Polym Sci 73:1231–1238
Li J, Zeng X, Kong D et al (2018) Polym Compos 39:858–868
Cheng X-W, Guan J-P, Tang R-C, Liu K-Q (2016) J Clean Prod 124:114–119
Dai X, Li P, Sui Y, Zhang C (2021) Eur Polym J 147:110319
Chang YL, Wang YZ, Ban DM, Yang B, Zhao GM (2004) Macromol Mater Eng 289:703–707
Gao L-P, Wang D-Y, Wang Y-Z, Wang J-S, Yang B (2008) Polym Degrad Stab 93:1308–1315
Zhang C, Huang JY, Liu SM, Zhao JQ (2011) Polym Adv Technol 22:1768–1777
Xiong YQ, Zhang XY, Liu J et al (2012) J Appl Polym Sci 125:1219–1225
Yang S, Zhang Q, Hu Y (2016) Polym Degrad Stab 133:358–366
Vothi H, Nguyen C, Pham LH, Hoang D, Kim J (2019) ACS Omega 4:17791–17797
Jeng R-J, Shau S-M, Lin J-J, Su W-C, Chiu Y-S (2002) Eur Polym J 38:683–693
Zhao Q, Chen C, Fan R, Yuan Y, Xing Y, Ma X (2017) J Fire Sci 35:99–117
Liu J, Dong C, Zhang Z, Sun H, Kong D, Lu Z (2020) Cellulose 27:9027–9043
Yuan Y, Yang H, Yu B et al (2016) Ind Eng Chem Res 55:10813–10822
Leu TS, Wang CS (2004) J Appl Polym Sci 92:410–417
Nguyen C, Kim J (2008) Polym Degrad Stab 93:1037–1043
Zheng Z, Liu BW, Ting Yang S, Cui X, Wang H (2015) Polym Compos 36:1606–1619
Wang D, Liu Q, Peng X et al (2021) Polym Degrad Stab 187:109544
Markwart JC, Battig A, Zimmermann L et al (2019) ACS Appl Polym Mater 1:1118
Battig A, Markwart JC, Wurm FR, Schartel B (2019) Polym Chem 10:4346–4358
Sim M-J, Cha S-H, Lee J-C (2021) Polym Test 100:107266
Lu Y, Wu C, Xu S (2018) Compos A Appl Sci Manuf 113:1–1
Huang Z, Wang Z (2021) Polym Degrad Stab 183:109425
Li M, Zhang J, Huang K, Li S, Jiang J, Xia J (2014) RSC Adv 4:63576–63585
Ye L, Li T, Hong L (2021) Mater Today Commun 26:102186
Gupta A, Lakshmp Y, Manivannan R, Victoria SN (2017) J Chil Chem Soc 62:3393–3398
Tan J, Liu B, Fu Q, Wang L, Xin J, Zhu X (2019) Polymers 11:779
Zhou J, Gui B, Qiao Y et al (2016) Fuel 166:526–532
Song L, Xuan S, Wang X, Hu Y (2012) Thermochim Acta 527:1–7
Zhan Z, Xu M, Li B (2015) Polym Degrad Stab 117:66–74
Yuan Y, Ma C, Shi Y, Song L, Hu Y, Hu W (2018) Mater Chem Phys 211:42–53
Fang F, Song P, Ran S, Guo Z, Wang H, Fang Z (2018) Compos Commun 10:97–102
Sun Z, Hou Y, Hu Y, Hu W (2018) Mater Chem Phys 214:154–164
Zhang P, Fan H, Tian S, Chen Y, Yan J (2016) RSC Adv 6:72409–72422
Wang C, Zhao J, Luo S, Yu X (2021) Macromol Res 29:582–588
Zhu Z-M, Wang L-X, Lin X-B, Dong L-P (2019) Polym Degrad Stab 169:108981
Hu X, Yang H, Jiang Y et al (2019) J Hazard Mater 379:120793
Zhong Y, Wu W, Lin X, Li M (2014) J Appl Polym Sci 131:40080
Wu DH, Zhao PH, Liu YQ, Liu XY, Wang XF (2014) J Appl Polym Sci, 131
Zhang T, Yan H, Shen L et al (2014) RSC Adv 4:48285
Xing W, Song L, Jie G, Lv X, Wang X, Hu Y (2011) Polym Adv Technol 22:2123–2129
Qian X, Song L, Bihe Y et al (2014) Mater Chem Phys 143:1243–1252
Chen X, Hu Y, Song L (2008) Polym Eng Sci 48:116–123
Ji S, Duan H, Chen Y, Guo D, Ma H (2020) Polymer 207:122917
Huo S, Wang J, Yang S et al (2018) Polym Adv Technol 29:497–506
Yang Y, Xiao D (2022) E-Polymers 22:430–444
Xue Z, Zhang W, Yan M, Liu J, Wang B, Xia Y (2017) RSC Adv 7:25253–25264
Liang S, Neisius M, Mispreuve H, Naescher R, Gaan S (2012) Polym Degrad Stab 97:2428–2440
Gaan S, Liang S, Mispreuve H, Perler H, Naescher R, Neisius M (2015) Polym Degrad Stab 113:180–188
Liang S, Hemberger P, Steglich M et al (2020) Chem A Eur J 26:10795–10800
Liang S, Hemberger P, Neisius NM et al (2015) Chem Eur J 21:1073–1080
Liang S, Hemberger P, Levalois-Grützmacher J, Grützmacher H, Gaan S (2017) Chem Eur J 23:5595–5601
Acknowledgements
The authors would like to thank the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A1071795).
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1F1A1071795).
Author information
Authors and Affiliations
Contributions
HHK: validation, formal analysis, investigation, project administration, writing—original draft. MJS: conceptualization, methodology, formal analysis, investigation, writing—original draft. SHC: conceptualization, formal analysis, writing—review & editing, Supervision, Funding acquisition. JChL: conceptualization, Methodology, Supervision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not Applicable.
Additional information
Handling Editor: Jaime Grunlan.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kim, HH., Sim, MJ., Lee, JC. et al. The effects of chemical structure for phosphorus-nitrogen flame retardants on flame retardant mechanisms. J Mater Sci 58, 6850–6864 (2023). https://doi.org/10.1007/s10853-023-08414-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-023-08414-6