
Abstract
Porous silicon (PSi) has become one of the current hotspots in lithium-ion anode materials research. However, due to their large volume change in the charge and discharge processes, which results in poor electrochemical performance, the material’s practical value is significantly impacted. In this study, PSi@poly(3,4-ethylenedioxythiophene) (PSi@PEDOT) composites were prepared by coating porous micron Si etched from the commercial Al–Si alloy with a conductive polymer synthesized via the in situ polymerization of 3,4-ethylenedioxythiophene (EDOT). The PEDOT shell not only ensures the conductive network in the composite material but also inhibits Si volume expansion. In addition, the pores inside the PSi material’s core layer provide space gaps to buffer Si volume expansion, which can effectively improve its cycling stability and rate performance. Electrochemical test results show that the PSi@PEDOT-2 composite with mass ratio of 1.5:1 exhibits the best cycling stability. The initial discharge capacity is 1338.62 mAh g−1 at a current density of 0.5 A g−1, and the postcapacity retention rate is 70.3% after 200 cycles. Further, the preparation method may be used as an approach for preparing high-performance Si-based anode materials.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
High energy density has become a critical performance measure for the global battery industry due to the rapid development of lithium-ion (Li-ion) batteries and the growing need for human activities [1,2,3,4]. However, the theoretical specific capacity of conventional graphite anodes (372 mAh g−1) is low [5, 6], which cannot satisfy the energy density required by large-scale equipment such as the new energy vehicles. Therefore, the development of high specific capacity and stable Li-ion anode materials is important [7, 8]. Moreover, Si has become the most promising next-generation anode material for Li-ion batteries [9, 10] due to its ultrahigh theoretical specific capacity (4200 mAh g−1) [11, 12] and low Li intercalation potential [13, 14]. However, because of Si’s poor conductivity and the huge volume changes [15, 16] that occur during charging and discharging, the active material smashes, falls off the current collector, and becomes inactive, resulting in poor battery cycle and rate performances, severely hindering the industrialization process of Si anode materials. Thus, effective techniques and approaches to increase the electrochemical performance of Si-based anode materials are urgently needed.
Researchers have investigated the electrochemical characteristics of Si in various ways in an attempt to tackle the difficulty created by its volume expansion. (1) Si has a nanosized [17, 18] and porous [19, 20] structure. Nanoscale Si particles are small in size, which can reduce their volume effect during charging and discharging, reduce particle spacing, and increase the ion transmission rate. After making the Si particles porous, the pore structures inside the Si particles can minimize the expansion stresses, allowing the Si structure to remain stable for a longer time; (2) the material’s electrochemical performance can be enhanced to an extent by reducing its size and porosity, but its electrochemical performance can be better improved if it is dispersed in a compounding material that cannot only conduct electricity but also buffer volume changes [21, 22].
Currently, nanosilicon is the frequently used Si, which exhibits higher electrochemical performance; however, its production cost is relatively high, thus causing difficulty in production expansion [23]. As an anode material, Si has poor conductivity [24,25,26]. However, it has recently attracted significant attention using conductive polymers to polymerize and improve its conductivity [27, 28]. Common conductive polymers are polythiophene [29], polyacetylene [30], polypyrrole [31], polyaniline [32], and many more. Poly(3,4-ethylenedioxythiophene) (PEDOT), for example, exhibit significant application value due to its advantages of high electrical conductivity, high light transmittance in the visible light region, good film formability, high stability under light and heat, and environmental friendliness [33]; thus, Li-ion batteries have been widely employed. As an electronic conductive polymer, PEDOT can improve charge-transfer reactions on the electrode surface and prevent excessive Si from continuously forming new solid electrolyte interphase (SEI) films with the electrolyte [34, 35]. Joseph et al. [36] synthesized Li+-PEDOT:PSS@CG film with outstanding electrolyte wettability, ionic conductivity, and interfacial characteristics. Zhang et al. [37] synthesized a ZnFe2O4-PEDOT composite material with a network of macroporous channels using PEDOT as the coating layer, and the cycle and rate performances were improved. The above results confirm that the use of PEDOT as a coating layer can improve the electrochemical performance of the negative electrode material. The PEDOT coating layer can be used as an effective buffer layer to minimize the material’s volume change during charging and discharging processes. In addition, as a conductive medium, PEDOT promotes the migration of electrons during charging and discharging, which improves the material’s ion migration speed as well as the battery’s rate performance.
Although nanosilicon exhibits good electrochemical performance, its production cost is high [38, 39]. In this study, commercialized aluminum–silicon (Al–Si) alloy and 3,4-ethylenedioxythiophene (EDOT) are prepared via in situ polymerization in an ice-water bath to synthesize a micron-scale PSi@PEDOT composite material with internal porosity and external core–shell structural features, which can increase the passage of ions and electrons while also absorbing the volume growth of internal Si, thus improving the material’s structural stability. Further, PEDOT as a coating layer can reduce the surface of porous silicon (PSi) in contact with the electrolyte, preventing excess Si from continuously generating new SEI membranes with the electrolyte, reducing the irreversible capacity loss caused by it, thus improving the material’s electrochemical performance.
Materials and methods
Preparation of PSi
Gradually add an appropriate amount of an Al–Si alloy to a 5% hydrochloric acid solution, stir at room temperature for 24 h and filter with suction. Further, wash the filtered cake with deionized water until the pH of the filtrate is approximately neutral, wash it three times with ethanol, and then, put it in a 60 °C vacuum drying oven for 12 h to obtain PSi.
Preparation of the PSi@PEDOT composite material
Add EDOT to the deionized water solution of polystyrene sulfonate (PSS) and mix it evenly. Transfer it to an ice-water bath, add PSi, and stir to disperse it evenly. The pH was changed to 2 by adding a 2-M HCl solution to the solution mixture. Then, a certain amount of initiator, (NH4)S2O4, was dissolved in 10-mL water and slowly added dropwise to the reaction mixture. After reacting in an ice-water bath for 6 h, it was transferred to room temperature to continue the reaction for 18 h, then centrifuged, and washed three times with deionized water to obtain a blue-black powdery PSi@PEDOT composite material, which was further dried in vacuum at 60 °C for 24 h.
Characterization
The morphology and microstructure of the samples were observed using a ZEISS SIGMA 300 tungsten filament scanning electron microscope (SEM) and FEI-TF20 transmission electron microscope (TEM). A Bruker-D8-Advance X-ray diffractometer (XRD) was used for the crystal structure test. A NETZSCH STA2500 thermal analyzer was used to perform thermogravimetric analysis (TGA) of the samples. The test conditions were as follows: In an air atmosphere, heating from room temperature (10 °C min−1) to 800 °C. The material’s specific surface and pore size distribution were investigated using the Best 3H-2000PS2 specific surface analyzer. To test and analyze the material’s surface composition, Thermo Scientific K-alpha X-ray photoelectron spectroscopy (XPS), with an excitation source of Al Kα rays was used. A Fourier-transform infrared spectrometer was used to determine the material’s functional groups. A Raman Thermo DXR microlaser was used to conduct Raman spectroscopy at an excitation wavelength of 532 nm to determine the material’s composition.
Electrochemical measurements
The prepared negative active material, conductive agent, and binder are weighed and dissolved in a suitable quantity of water in a mass ratio of 7:1:2, mixed uniformly, coated with copper foil, and dried in a vacuum drying oven. Further, the battery is assembled in a high-purity argon glove box with the prepared negative electrode sheet and tested. The Xinwei BTS-5V10 mA tester is used to conduct a constant current charging and discharging test on the battery. Notably, the test temperature is 25 °C; voltage range is 0.01–1.5 V (vs. Li/Li+); formation current density is 0.1 A g−1; circulating current density is 0.5 A g−1; and rate current densities are 0.2, 0.4, 0.8, 1, 2, and 4 A g−1 and finally back to 0.2 A g−1. Cyclic voltammetry (CV) and impedance (EIS) tests are conducted using a multichannel electrochemical workstation. The CV test scans are performed at a rate of 0.1 mV s−1 and voltage range of 0.01–1.5 V. The EIS test frequency range is 0.01–100,000 Hz, and the test AC perturbation amplitude is 5 mV.
Results and discussion
Synthesis and structural characterization of PSi@PEDOT
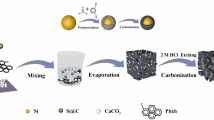
Figure 1a is a schematic of the synthesis path of the PSi@PEDOT composite material. There are mainly four steps in the whole process. First, use hydrochloric acid to etch the Al–Si alloy to prepare PSi and further disperse the EDOT and PSi uniformly using ultrasonic stirring. Finally, add an initiator to the mixed solution to initiate in situ polymerization of EDOT. The concentration and polymerization of EDOT can be controlled, and the coating thickness can be adjusted (by varying time). Figure 1b–j shows the SEM image of the sample PSi, PSi@PEDOT composite material, and elemental mappings (EDS) distribution map of PSi@PEDOT; the PSi remained spherical after being coated with PEDOT. Figure 2b shows that there are more holes in the spherical PSi. Figure 2c–d shows the SEM image of PEDOT-coated PSi. Because of the PEDOT coating, the material’s surface becomes smoother. Figure 2f–j represents the distribution map of Si, Al, C, O, and S in the yellow box (Fig. 2e). These five elements are observed to be uniformly distributed.

a Schematic illustration of synthesis route from PSi to PSi@PEDOT-2; b SEM images of (b) PSi; c–d PSi@PEDOT-2; f–j elemental mapping of PSi@PEDOT-2 in the yellow box in (e)

a, b, and d TEM images of PSi@PEDOT-2 and c SAED images of PSi@PEDOT
To further analyze the structure of the PSi@PEDOT composite material, TEM tests were conducted. Figure 2 shows the TEM image of PSi@PEDOT-2 as well as its internal structure (Fig. 3c), which is a selected area electron diffraction (SAED) map of the composite material, corresponding to the Si(111), Si(220), and Si(311) crystal planes. Figure 3d shows the lattice fringes of Si. The spacing is approximately 0.31 nm, corresponding to the (111) crystal plane of the crystalline Si. In addition, the interface of PEDOT-coated Si can be observed, with a coating thickness of approximately 3–4 nm.

a XRD patterns of PEDOT, Al–Si, PSi, and PSi@PEDOT-2; b FT-IR spectra of PEDOT, Al–Si, PSi, and PSi@PEDOT-2; c TGA thermogravimetric curves of PSi and PSi@PEDOT-2; and d N2 adsorption and desorption isotherm of PSi and PSi@PEDOT-2 (the inset: pore size distribution)
Figure 3a shows the XRD pattern of PSi@PEDOT-2, PSi, the Al–Si alloy, and PEDOT. The diffraction peaks of Al and Si (JCPDF77-2018) are observed in the pattern of the Al–Si alloy, implying that Si in the Al–Si alloy is crystalline. Only the diffraction peaks of crystalline Si appear in the spectra of PSi and PSi@PEDOT-2, indicating that the Al has been eliminated after acid treatment and that the crystalline state of the Si remains unchanged after the PEDOT coating and no other impurity phases exist. PEDOT has only a weak broad peak around 2θ = 24°, indicating the amorphous nature of the coated PEDOT. The diffraction peak of PSi@PEDOT-2 is weaker than that of PSi, which may be caused by the coating of amorphous PEDOT.
Fourier-transform infrared spectroscopy (FT-IR) spectroscopy is used to further understand the chemical structure of composite materials. Figure 3b shows the infrared spectra of PSi@PEDOT-2, PSi, the Al–Si alloy, and PEDOT. An additional peak at 3448 cm−1 corresponds to the –OH group in the absorption water at 1643, 1402, and 1337 cm−1. The peaks represent the C=C and C–C stretching vibrations of PEDOT on the thiophene ring. The representative peaks at 1200 and 1072 cm−1 are attributed to C–O–C tensile vibrations. The peak at 984 cm−1 corresponds to the vibration of the C–S bond in the thiophene ring. In addition, the PEDOT characteristic peaks can be observed in the PSi@PEDOT-2 composite FT-IR spectrum. These FT-IR results indicate that PEDOT was successfully polymerized on PSi.
The Si content of PSi@PEDOT composite material can be estimated based on the mass-loss rate of its thermogravimetric curve. The PSi@PEDOT-2 composite material has obvious mass loss at 300–500 °C (Fig. 3c), which is determined by the decomposition of PEDOT, and after 500 °C, the Si in the composite material is oxidized to SiOx, resulting in a slight increase in the composite material’s quality. Therefore, the mass loss in the composite material may be attributed to the polymer content, whereas the remaining quality is almost Si, that is, the content of Si in the PSi@PEDOT-2 composite is 77.5%, whereas that of PEDOT is 22.5%.
To verify the change in the material’s pore structure before and after the coating, BET tests were conducted on PSi and PSi@PEDOT-2. Figure 3d depicts the N2 adsorption and desorption isotherms as well as the pore size distribution curves. The N2 adsorption capacity of PSi abruptly increases when p/p0 is 0.8–1.0, resulting in an obvious hysteresis loop, which indicates numerous narrow crack holes in the PSi; the pore size is approximately 1–2 nm, PEDOT may be deposited on the PSi surface, obstructing some of the surface’s pores and reducing its specific surface area. It helps prevent more Si from reacting with the electrolyte to form an SEI film.
Surface characteristics of the PSi@PEDOT electrode
The elements on the surface of the PSi@PEDOT-2 composite material were characterized using XPS. The full spectrum of Fig. 4a shows that there are five elements of Si, Al, C, O, and S on the composite material’s surface. Figure 4b–d corresponds to the XPS spectra of Si 2p, S 2p, and Al 2p respectively. The characteristic peak at the binding energy of 98.5 eV corresponds to the Si–Si bond, and the peak at the binding energy of 102–104.5 eV is SiOx. The characteristic peak corresponds to the Si–O–Si bond, which may be because the Si surface of the PSi@PEDOT-2 composite material is slightly oxidized to form SiOx during the production process. The main peaks with binding energies of 164.7 and 166.0 eV correspond to S 2p3/2 and S 2p1/2 of the C–S–C covalent bond, respectively, whereas the two small peaks at 167.5 and 168.8 eV are attributed to the C–S(O)2–C of SO2. In addition, the weak characteristic peak of Al 2p is observed at the binding energy of 74.7 eV, indicating that there is still a small amount of Al in PSi, and the presence of a small amount of Al is conducive to improving the ionic conductivity of the material, thereby enhancing the reaction kinetics of Li deintercalation.

XPS spectra of the PSi@PEDOT-2 primitive electrode. a Elements complete spectra, b Si 2p spectra, c S 2p spectra, and d Al 2p spectra
Electrochemical performance of the PSi@PEDOT electrodes
The PSi@PEDOT-2 composite material’s electrochemical performance was studied via CV. Figure 5a shows that the strong reduction peak between 0 and 0.2 V corresponds to the conversion of crystalline Si to LixSi. In the subsequent cycle, the oxidation peak between 0.3 and 0.6 V corresponds to the LixSi delithiation reaction. In the PSi's second, third, and fourth CV curves, the reduction and oxidation peaks gradually increase (Fig. S2a), indicating a low PSi cycle reversibility. This is because the Si’s structure is damaged due to expansion, making the damaged Si unable to insert and release Li. The PSi@PEDOT-2 material’s CV curve in Fig. 5a increases with the overlap of the curve as the cycle progresses, indicating that the material’s cycle performance tends to be stable.

a Cyclic voltammetry curves of PSi@PEDOT from cycles 1–4 at a scanning rate 0.1 mV s−1; b charge/discharge curves of PSi@PEDOT electrodes; c cycle performance of PSi and PSi@PEDOT electrodes; d rate performance of PSi and PSi@PEDOT electrodes; e EIS spectra of PSi and PSi@PEDOT
Figure 5b shows the PSi@PEDOT-2 composite material’s charge and discharge curve. The first three circles are the formation charge and discharge curves (current density is 0.1A g−1), whereas the remaining three curves are cycles (0.5A g−1) of the charge and discharge curves of different turns, the first discharge capacity is 2553.5 mAh g−1, with initial Coulombic efficiency of 68.0%, the creation of the SEI film is indicated on the first discharge (lithiation) curve by an angled potential plateau between 1.1 and 0.2 V. The charge and discharge curves gradually overlap after several cycles, indicating that the cycle performance is gradually stable. The PSi@PEDOT composite materials with different Si contents have a minimal difference in the charge–discharge specific capacity at the first cycle, but as the reaction progresses, the capacity-retention rate of PSi@PEDOT-2 is observed to be relatively stable, whereas that of other materials decreases very quickly (Fig. S3). Indicating that an appropriate amount of PEDOT coating can maintain the stability of the material structure and prevent the reaction of excessive active materials with the electrolyte to form an SEI film, which is a major cause of irreversible capacity loss.
The effects of different raw material ratios on the PSi@PEDOT composite materials’ cycle performance were investigated. The cycle performance tests were conducted on four materials: PSi, PSi@PEDOT-1, PSi@PEDOT-2, and PSi@PEDOT-3 (Fig. 5c), and 200 charge–discharge cycles were performed at a current density of 0.5 A g−1. The specific capacity of the PSi@PEDOT-2 composite material, among others, decreases from 1254 to 878 mAh g−1, the capacity-retention rate can reach 70%, and the coulombic efficiency remains above 98.1%. Under the same test conditions, the specific capacity of PSi decreases from 1199 to 215 mAh g−1 after 100 cycles and the capacity-retention rate after 200 cycles is only 10.7%. This can be because the PSi@PEDOT-2 composite material can effectively buffer Si’s volume expansion and maintain the overall structure’s stability, thereby significantly improving the electrochemical cycle’s stability. The ability to maintain the structural integrity of the conductive network can also be verified by the better rate of performance of PSi@PEDOT-2. Figure 5d shows the PSi@PEDOT-2 composite material’s charging specific capacity decreases from 1140 to 313 mAh g−1 when the current density rises from 0.2 to 4 A g−1. However, when the current density returns to 0.2 A g−1, its specific capacity can rapidly increase to 1104 mAh g−1. Although the initial capacity of PSi at 0.2 A g−1 is higher than that of the PSi@PEDOT-2 composite material, it decreases rapidly in subsequent cycles. When it reaches 2 A g−1, the specific capacity is only 1 mAh g−1, indicating that Si is under high current density. The deintercalation of Li ions is nearly impossible, the conductivity of the PEDOT-coated PSi@PEDOT-2 composite material is enhanced, and the structure can remain stable under high current, indicating good rate performance.
Figure 5e shows the AC impedance diagram of the PSi and PSi@PEDOT electrodes of different ratios after 200 cycles. The semicircle diameter in the high-frequency zone can be used to quantify the charge that transfers impedance at the electrode interface. The semicircle in the high-frequency zone represents the charge-transfer resistance. Thus, the smaller the semicircle diameter, the smaller the charge-transfer resistance. Figure 5e also shows that the diameter of the PSi@PEDOT-2 electrode high-frequency semicircle is smaller than that of other electrodes after 200 cycles, indicating that the PSi@PEDOT-2 electrode’s charge-transfer resistance is smaller and the ion conductivity is higher; the conductivity results of the PSi and PSi@PEDOT electrodes of different ratios can also be calculated by using the sheet resistance measured by the four-probe method, as shown in Table S1, the conductivity of the PSi@PEDOT electrode is greater than that of the PSi electrode, thus, its electrochemical performance is better.
SEM images of electrode sheet
In order to prove that the PSi@PEDOT composite material can maintain the stability of the structure and inhibit the expansion of porous silicon, the morphology of the PSi and PSi@PEDOT-2 electrode sheets before and after lithium insertion and before and after the cycle were observed by SEM. As shown in Fig. 6, in the state where the pole pieces are filled with lithium, the particles of PSi (Fig. 6b) are significantly larger than those of PSi@PEDOT-2 (Fig. 6f). After 200 cycles of the PSi pole pieces, the material on the film has wide cracks caused by expansion (Fig. 6d), while the material of the PSi@PEDOT-2 pole piece has no obvious cracks, and the morphology and structure are maintained well (Fig. 6h). The above phenomenon shows that PSi@PEDOT composite material can effectively buffer the volume expansion of silicon.

SEM images of electrode sheet a PSi electrode sheet before lithium insertion; b PSi electrode sheet after lithium insertion; e PSi@PEDOT-2 electrode sheet before lithium insertion; f PSi@PEDOT-2 electrode sheet after lithium insertion; c PSi electrode sheet before cycling; d PSi electrode sheet after 200 cycles; g PSi@PEDOT-2 electrode sheet before cycling; h PSi@PEDOT-2 electrode sheet after 200 cycles
Conclusions
We successfully developed a novel approach for fabricating micron Si-based anodes coated with a conductive polymer that demonstrates high electrochemical performances. It is crucial to understand that PEDOT can provide a high conductivity channel for electron transmission and the porous structure of PSi@PEDOT outer core and shell can effectively reduce Si’s volume-expansion rate. PSi@PEDOT-2 with a Si mass fraction of 77.5% demonstrates the best cycle stability with a capacity-retention rate of 70% after 200 cycles at 0.5 A g−1. Thus, PSi@PEDOT will be a potential alternative for Li-ion Si-based anodes in the future due to its high electrochemical performance and simple economic preparation technique.
References
Ullah A, Majid A, Rani N (2018) A review on first principles based studies for improvement of cathode material of lithium ion batteries. J Energy Chem 27(1):219–237
Bai J, Chen X, Olsson E (2020) Synthesis of Bi2S3/carbon nanocomposites as anode materials for lithium-ion batteries. J Mater Sci Technol 50:91–102
Yao Y, Liu N, Mcdowell MT (2012) Improving the cycling stability of silicon nanowire anodes with conducting polymer coatings. Energy Environ Sci 5(7):7927–7930
Dai X, Lei SL, Liu J, Shang ZT, Zhong SW, Li XC (2021) Promoting the energy density of lithium-ion capacitor by coupling the pore-size and nitrogen content in capacitive carbon cathode. J Power Sources 498:229912.
Wu W, Kang Y, Wang M (2020) An ultrahigh-areal-capacity SiOx negative electrode for lithium ion batteries. J Power Sources 464:228–244
Li D, Zhang X, Wang Y (2021) Adjusting ash content of char to enhance lithium storage performance of rice husk-based SiO2/C. J Alloys Compd 854:156986.
Liu XJ, Iqbal A, Ali N, Qi RR, Qian XF (2020) Ion-crosslinking promoted high performance Si/PEDOT:PSS electrodes: the importance of cations’ ionic potential and softness parameters. ACS Appl Mater Interfaces 12(17):19431–19438
Li XM, Feng ZX, Zai JT, Ma ZF, Qian XF (2018) Incorporation of Co into MoS2/graphene nanocomposites: one effective way to enhance the cycling stability of Li/Na storage—sciencedirect. J Power Sources 373:103–109
Wei R, Xu R, Zhang K (2021) Biological enzyme treatment of starch-based lithium-ion battery silicon-carbon composite. Nanotechnology 32(4):045605.
Yang Y, Lu Z, Xia J (2020) Crystalline and amorphous carbon double-modified silicon anode: towards large-scale production and superior lithium storage performance. Chem Eng Sci 229:116054.
Hong Z, Ning TB, Wz A (2021) Novel synthesis of Silicon/Carbon nanotubes microspheres as anode additives through chemical vapor deposition in fluidized bed reactors. Scr Mater 192:49–54
Cheng X, Na R, Wang X (2019) Si nanoparticles embedded in 3D carbon framework constructed by sulfur-doped carbon fibers and graphene for anode in lithium-ion battery. Inorg Chem Front 6(8):1996–2003
Xu YH, Yin GP, Cheng XQ (2011) Enhanced lithium storage performance of silicon anode via fabricating into sandwich electrode. Electrochim Acta 56(11):4403–4407
Tang F, Jiang T, Tan Y et al (2021) Preparation and electrochemical performance of silicon@graphene aerogel composites for lithium-ion batteries. J Alloys Compd 854:157135.
Xu R, Wang G, Zhou T (2017) Rational design of Si@carbon with robust hierarchically porous custard-apple-like structure to boost lithium storage. Nano Energy 39:253–261
Park E, Kim J, Chung DJ (2016) Si/SiOx-Conductive polymer core-shell nanospheres with an improved conducting path preservation for lithium-ion battery. Chemsuschem 9(19):2754–2758
Chen D, Tao C, Chen T (2021) Pomegranate-like silicon-based anodes self-assembled by hollow-structured Si/void@C nanoparticles for li-ion batteries with high performances. Nanotechnology 32(9):095402.
An W, Gao B, Mei S (2019) Scalable synthesis of ant-nest-like bulk porous silicon for high-performance lithium-ion battery anodes. Nature Commun 10(1):1447
Wang F, Sun L, Zi W (2019) Solution synthesis of porous silicon particles as an anode material for lithium ion batteries. Chemistry 25(38):9071–9077
Hwang C, Lee K, Um HD (2017) Conductive and porous silicon nanowire anodes for lithium-ion batteries. J Electrochem Soc 164(7):A1564–A1568
Liu XJ, Xu ZX, Iqbal A, Chen M, Low CTJ, Qi RR, Zai JT, Qian XF (2021) Chemical coupled PEDOT:PSS/Si electrode: suppressed electrolyte consumption enables long-term stability. Nano-Micro Lett 13(3):12
Li B, Li SX, Jin Y, Zai JT, Chen M, Ali N, Zhan P, Huang Y, Qian XF (2018) Porous Si@C ball-in-ball hollow spheres for lithium-ion capacitors with improved energy and power densities. J Mater Chem A 6(42):21098–21103
Zhao T, Zhu D, Li W (2019) Novel design and synthesis of carbon-coated porous silicon particles as high-performance lithium-ion battery anodes. J Power Sources 439:227027.
Troegel D, Amann M, Haufe S (2014) Silicon/Carbon-Composites as anode materials for lithium ion. Batteries 2(1):149–198
Zhou Y, Yang Y, Hou G (2020) Stress-relieving defects enable ultra-stable silicon anode for li-ion storage. Nano Energy 70:104568.
Tao W, Wang P, You Y (2019) Strategies for improving the storage performance of silicon-based anodes in lithium-ion batteries. Nano Res 12(8):1739–1749
Mcgraw M, Kolla P, Cook R (2016) One-step solid-state in-situ thermal polymerization of Silicon-PEDOT nanocomposites for the application in lithium-ion battery anodes. Polymer 99:488–495
Zhou X, Liu Y, Du C (2018) Polyaniline-encapsulated silicon on three-dimensional carbon nanotubes foam with enhanced electrochemical performance for lithium-ion batteries. J Power Sources 381(31):156–163
Na RQ, Krysten M, Zhang GY (2019) Electrically conductive shell-protective layer capping on the silicon surface as the anode material for high-performance lithium-ion batteries. ACS Appl Mater Interfaces 11(43):40034–40042
Liao Y, Liang K, Ren Y (2020) Fabrication of SiOx-G/PAA-PANi/Graphene composite with special cross-doped conductive hydrogels as anode materials for lithium ion batteries. Front Chem 8:96
Wu C Y, Kuo P H, Duh J G (2021) Reviving of silicon waste with N-doped carbon Core-Shell structure prepared by vapor deposition polymerization of Polypyrrole applied in Lithium-ion battery. Surf Coat Technol 421:127418.
Liu H, Zou Y, Huang L (2018) Enhanced electrochemical performance of sandwich-structured polyaniline-wrapped silicon oxide/carbon nanotubes for lithium-ion batteries. Appl Surf Sci 442(1):204–212
Rajesh M, Manikandan R, Kim B C (2020) Electrochemical polymerization of chloride doped PEDOT hierarchical porous nanostructure on graphite as a potential electrode for high performance supercapacitor. Electrochim Acta 354:136669.
Liu XJ, Zai JT, Iqbal A, Chen M, Ali N, Qi RR, Qian XF (2020) Glycerol-crosslinked PEDOT:PSS as bifunctional binder for Si anodes: Improved interfacial compatibility and conductivity. J Colloid Interface Sci 565:270–277
Chen M, Li B, Liu XJ, Zhou L, Yao L, Zai JT, Qian XF, Yu XB (2018) Boron-doped porous Si anode materials with high initial coulombic efficiency and long cycling stability. J Mater Chem A 6(7):3022–3027
Hareendrakrishnakumar H, Chulliyote R, Joseph MG (2021) Ion-selective PEDOT:PSS-decorated separator as a potential polysulfide immobilizer for lithium-sulfur batteries. Ionics 27(3):1087–1099
Zhang M, Liu Y, Zhu H (2020) Hierarchical bead chain ZnFe2O4-PEDOT composites with enhanced Li-ion storage properties as anode materials for lithium-ion batteries. Appl Surf Sci 529:147078.
Fan Z, Zheng S, He S (2020) Preparation of micron Si@C anodes for lithium ion battery by recycling the lamellar submicron silicon in the kerf slurry waste from photovoltaic industry. Diamond Relat Mater 107:107898.
Wan W, Y Mai, D Guo (2021) A novel sol-gel process to encapsulate micron silicon with a uniformly Ni-doped graphite carbon layer by coupling for use in lithium ion batteries. Synth Met 274(1):116717.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51862013) and Science and Technology Project of Jiangxi Education Department (GJJ200810).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Handling Editor: Mark Bissett.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, R., Chen, Y., Ding, N. et al. Studies on the preparation and electrochemical performance of PSi@Poly(3, 4-ethylenedioxythiophene) composites as anode materials for lithium-ion batteries. J Mater Sci 57, 4323–4333 (2022). https://doi.org/10.1007/s10853-022-06922-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-06922-5