
Abstract
To effectively decompose the organic dyes in wastewater, a novel all-solid state Z-scheme In2O3/WO3 heterostructured photocatalyst was successfully prepared by loading In2O3 nanoparticles onto WO3 nano-needles through a two-step hydrothermal–solvothermal method. The phase structures, morphologies, chemical compositions, and optical adsorption properties of these photocatalysts were characterized in detail. In the Z-scheme photocatalytic system, the built-in internal electric field can accelerate the recombination of useless photo-generated holes on the VB of In2O3 and electrons on the CB of WO3. The retaining photo-generated charge carriers on the CB of In2O3 and VB of WO3 possess strong redox ability. Therefore, the In2O3/WO3 heterogeneous photocatalysts exhibited remarkably improved photocatalytic activity toward degradation of organic dyes and tetracycline hydrochloride compared to pure WO3 and In2O3 semiconductor materials under visible-light irradiation. The recycling experiments showed that Z-scheme In2O3/WO3 heterogeneous photocatalyst could still degrade 86.6% of methylene blue and 86.4% of rhodamine B even after three cycles, confirming its high photo-stability. The trapping experiments demonstrated that photo-generated holes and ·O2− were the predominant active species for photocatalytic degradation of organic dyes. Based on the experimental results, a possible photocatalytic mechanism of Z-scheme In2O3/WO3 heterostructure was proposed. This investigation provided a novel approach for construction of efficient heterostructured photocatalysts for wastewater purification.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the first synthetic dye mauveine came out in 1856, the dyestuff industry has developed rapidly and offered various dyes for textile, paper-making, and leather industries. However, the use of synthetic dye also brings about serious environmental problems, because approximately 10–15% of dyes cannot be utilized during dyeing process and released into natural water system as colored effluents [1]. The organic dye residues in environment have been proved to possess negative impact on aquatic life as well as human health [2,3,4,5,6,7]. Differing from natural dyes, synthetic dyes usually have low biodegradability due to their stable chemical structure and cannot be easily removed by conventional wastewater treatment methods. Therefore, it is highly urgent to develop efficient methods and techniques for the elimination of synthetic dyes from dyeing effluent.
Recently, semiconductor photocatalysis has received great attention as one of the most desirable and environment-friendly methods to decompose organic dyes using renewable solar light energy [8,9,10,11]. So far, many photocatalyst nanoparticles have been developed for wastewater treatment, such TiO2, ZnO, BiVO4, g-C3N4, Ag3PO4, and BiOX(X = Cl, Br, I) [12]. Compared with other treatment methods, photocatalysis technology has its distinct advantages, such as nontoxicity, easy operation, mild reaction conditions, and low-cost [13, 14]. However, the photocatalytic activities of single photocatalysts are usually inhibited due to their high recombination rate of photo-generated charge carriers and their low utilization of visible light energy. The construction of semiconductor heterostructured photocatalysts is proposed as a feasible and promising strategy to address the above limitations [15,16,17]. Among the proposed heterojunction systems, all-solid state Z-scheme heterostructures have been proven to be one of the most effective ways for fabricating high efficient photocatalysts due to their relatively high redox potential [18,19,20,21]. The Z-scheme photocatalytic system is usually composed of oxidation photocatalyst (PS I) and reduction photocatalyst (PS II). For the purpose of maximizing performances of the Z-scheme system, PS I and PS II should possess low valence band (VB) position and high conduction band (CB) position, respectively.
Tungsten oxide (WO3) is an important metal oxide semiconductor photocatalyst with relatively narrow band gap (2.7 eV) and high photo-stability. Generally, its conduction band (CB) position is about 0.74 eV, and the valence band (VB) position is about 3.44 eV [22]. The highly positive VB level of WO3 endows photo-generated holes with a strong oxidative capability. However, the photocatalytic activity of pure WO3 under visible-light irradiation is actually unsatisfactory due to its rapid recombination of photo-generated charge carriers and their sluggish migration. It is generally known that the morphology of photocatalyst plays a pivotal role in determining its photocatalytic efficiency. According to previous literature [23,24,25,26], WO3 possesses various morphologies such as nanoparticles, nanoflowers, nanosheets, and nanorods. Among various WO3 nanostructures, one-dimensional (1D) WO3 is considered as the optimal morphology for photocatalysis due to the high surface-to-volume ratios and shorter migration paths of photo-generated charge carriers. Moreover, coupling WO3 with other semiconductor photocatalysts, such as TiO2/WO3 [27], WO3/g-C3N4 [28], Ag3PO4/WO3 [29], WO3/Ag2CrO4 [30], to form heterojunctions has been reported as an effective strategy of promoting the separation of photo-generated electron–hole pairs. WO3 with more positive VB position is well suited as the PS I in Z-scheme heterogeneous photocatalyst. In this case, to seek a semiconductor photocatalyst with appropriate CB potential is important for the construction of heterojunction photocatalytic system containing WO3. Indium oxide (In2O3), with a band gap of 2.8 eV, is a well-known n-type semiconductor material and has been widely coupled with other photocatalysts to construct heterogeneous photocatalytic structure [31,32,33,34]. The CB and VB potentials of In2O3 are -0.63 eV and 2.17 eV, respectively, vs. normal hydrogen electrode (NHE) [35]. The more negative CB potential makes In2O3 a feasible candidate for the construction of Z-scheme photocatalytic system as the reduction photocatalyst.
Herein, an all-solid state Z-scheme In2O3/WO3 photocatalyst was fabricated by depositing In2O3 nanoparticles on 1D WO3 nano-needles via a facile two-step hydrothermal–solvothermal route. The photocatalytic activity of In2O3/WO3 was evaluated by the degradation of MB, RhB, and TC under visible-light irradiation. The as-prepared In2O3/WO3 composite photocatalyst exhibited excellent photocatalytic performance under visible-light irradiation, in comparison with pure WO3 and In2O3. The prepared samples were systematically characterized in terms of phase structures and morphologies, chemical bonding states, optical properties, and electrochemical properties. The radicals trapping experiments were conducted to detect the main active species during the photocatalytic reaction. The degradation intermediates of organic dyes were analyzed by HPLC–MS. Subsequently, a possible photocatalytic mechanism for degradation of organic dyes was also proposed.
Experimental section
Materials
Indium (III) nitrate hydrate (In(NO3)3·4.5H2O), D(+)-glucose monohydrate (C6H12O6·H2O), urea (CO(NH2)2), tert-butyl (t-BuOH), ammonium oxalate (AO), and benzoquinone (BQ) were purchased from the Sinopharm Chemical Reagent Co., Ltd. Sodium tungstate dihydrate (Na2WO4·2H2O), rhodamine B (RhB), methylene blue (MB), and tetracycline hydrochloride (TC) were obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Sodium chloride (NaCl), ethanol absolute (CH3CH2(OH)), and hydrochloric acid (HCl) were purchased from Sigma-Aldrich. All of the above chemicals were used without further purification. Deionized (DI) water was used throughout the experiments.
Preparation of WO3 nano-needle photocatalysts
WO3 photocatalysts were prepared by hydrothermal method. In a typical procedure, 1 g of Na2WO4·2H2O and 0.2 g of NaCl were added into 25 mL of DI water and stirred magnetically for 3 h. Then, the pH value of the above suspension was adjusted to 2.0 using HCl solution. After magnetic stirring for another 2 h, the mixture was transferred into a 50-mL Teflon-lined stainless autoclave, sealed, and maintained at 200 °C for 20 h in a muffle furnace. The precipitates were collected out and washed with DI water and ethanol for three times, respectively. Finally, WO3 photocatalysts were obtained after drying at 60 °C for 8 h in a vacuum oven.
Preparation of Z-scheme In2O3/WO3 photocatalyst
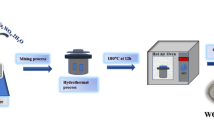
The Z-scheme In2O3/WO3 (In/W) heterogeneous photocatalysts were prepared via a solvothermal method. Typically, 129 mg of WO3 photocatalysts was added into the mixture of diethylene glycol (20 mL) and DI water (8 mL). The WO3 dispersion was treated using ultrasonic for 20 min and stirred magnetically for 180 min. Subsequently, a certain amount of In(NO3)3·4.5H2O was added into the above dispersion and stirred for 120 min. Then, 2.5 g of CO(NH2)2 was added into the mixture solution. After stirring for another 60 min, the obtained solution was transferred into a 50-mL Teflon-lined stainless autoclave and maintained at 200 °C for 18 h. As the autoclave was naturally cooled to room temperature, the obtained products were collected by centrifugation and washed thoroughly with ethanol and DI water. Finally, the composite photocatalysts were obtained after drying at 60 °C for 5 h in a vacuum oven. The obtained samples are labeled as In/W-x (x = 1, 2, 3), which represents the different mass fractions (5%, 10%, and 15%) of In2O3 in composite photocatalysts, respectively. For comparison, the pure In2O3 photocatalyst nanoparticles were also prepared by the same method without adding WO3 photocatalysts (Fig. 1).

Schematic illustration of the preparation process for In/W-x
Characterization
The crystalline structures of the prepared composite photocatalysts were acquired by X-ray powder diffraction (XRD, Rigaku Ltd., Japan) in the range of 10–80° (2θ), using Cu Kα (α = 1.54056 Å) radiation. The morphology and microstructure of samples were observed using a scanning electron microscope (SEM) (HITACHI S-4800, Japan) with an energy-dispersive X-ray spectroscopy (EDS) and a transmission electron microscope (TEM) (JEOL JEM-2100, Japan). The chemical composition of the samples was determined by X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250XI). All the binding energies were referenced to the C 1 s peak at 284.8 eV. The UV–Vis diffuse reflectance spectroscopy (UV–Vis DRS) was recorded using a UV–Vis spectrophotometer (U-3900H, HITACHI, Japan) equipped with an integrating sphere attachment. Photoluminescence (PL) spectra were tested on an Edinburgh FS5 Fluorescence Spectrometer. The specific surface areas of the photocatalysts were investigated at 77 K by Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption methods with a Micromeritics Tristar II 3020 system. The transient photocurrent and electrochemical impedance spectroscopy (EIS) were performed on an electrochemical workstation (CHI760D Chenhua Instrument Company) with a three-electrode system. Electron spin resonance (ESR) spectroscopy was measured on an electron paramagnetic resonance spectrometer (Bruker A300, Germany) using spin capture reagent DMPO in water and methyl alcohol.
Photocatalytic activity evaluation
The photocatalytic activity of prepared photocatalyst was evaluated by the photocatalytic degradation of organic dyes (MB and RhB) and antibiotics TC under the illumination of a 500 W Xe lamp with a 420-nm cutoff filter. All of the photocatalytic experiments were carried out at room temperature and atmospheric pressure. Typically, a certain amount of photocatalysts were dispersed homogeneously in 50 mL of MB (5 mg/L), RhB (25 mg/L), and TC (20 mg/L) aqueous solution. Before irradiation, the suspensions were magnetically stirred in the dark for 30 min to establish adsorption–desorption equilibrium between the dyes and photocatalysts. Subsequently, the Xe lamp was turned on and cooled by a quartz cold trap throughout the photocatalytic experiments. At a periodic interval, constant amounts of suspensions were taken out and filtered to remove the photocatalyst powders. To investigate the cycling stability, the photocatalysts after photocatalytic reaction were separated from dispersions by centrifugation and washed with ethanol and deionized water for three times. The dried photocatalysts were reused in the fresh dye solutions for the next cycle. The concentrations of MB, RhB, and TC filtrates were determined by spectrophotometry using a UV–Vis spectrophotometer (U-3900H, HITACHI, Japan). The photocatalytic degradation rate of the dye was calculated according to the following Eq. (1):
where R is the dye degradation rate; C0 is the initial concentration of dye solution; Ct is the concentration of dye solution at a given irradiation time t. The total organic carbon (TOC) during photocatalytic degradation of organic dyes by different samples was determined to evaluate the mineralization ability of the corresponding photocatalysts.
Free radicals trapping experiments were also carried out to determine the main active species in the photocatalytic degradation of organic dyes. Three different scavengers, t-BuOH, AO, and BQ, were chosen for capturing ·OH, h+, and ·O2−, respectively. In every experiment, 0.5 mL of scavenger solutions (1 mmol/L) was added into the reaction mixtures before light irradiation.
The degradation intermediates of two organic dyes were analyzed by HPLC–MS (Thermo Scientific, USA) with C18 column (100 mm × 4.6 mm i.d., 5 μm). The mixture of two solvents (acetonitrile: 0.1% Formic acid solution = 60:40, v/v) was used as a mobile phase. The flow rate was 0.6 mL/min and the injection volume was 5 μL. The column temperature was maintained at 30 °C.
Results and discussion
XRD analysis
The crystalline structures of the as-prepared samples were characterized by XRD analysis. As shown in Fig. 2, all the diffraction peaks of WO3 photocatalyst could be ascribed to the standard XRD patterns of monoclinic phase WO3 (JCPDS20-1324) with the major peaks at 2θ = 23.08°, 23.71°, 24.09°, 26.59°, 28.77°, 33.64°, 34.02°, 41.52°, 47.23°, 48.43°, 50.49°, 53.68°, and 54.30°, corresponding to the diffractions of the (001), (020), (200), (120), (111), (201), (220), (221), (002), (040), (112), (202), and (041) crystal planes, respectively [22]. The XRD patterns of In2O3 exhibited several peaks at 21.50°, 30.58°, 35.47°, 45.69°, 51.04°, and 60.68°, which were attributed to the diffraction peaks of the (211), (222), (400), (431), (440), and (622) crystal planes (JCPDS71-2195), respectively [36]. For the composite photocatalyst In2O3/WO3, each of the diffraction peaks of WO3 could be observed in their XRD patterns, indicating that the crystalline structure of WO3 photocatalyst was not affected by the introduction of In2O3. Moreover, the strongest characteristic diffraction peak of In2O3 at 30.58° could also be found in the composite photocatalysts and its intensity increased gradually with the increasing contents. The other characteristic diffraction peaks of In2O3 could not be observed obviously owing to the low content. The XRD results confirmed that In2O3 nano-photocatalysts were successfully loaded on the surface of WO3 nano-needle photocatalysts.

XRD patterns of WO3, In2O3, and In/W-x photocatalysts
Morphological characterization
The morphologies and microstructures of the samples were investigated by SEM and HRTEM. Figure 3a shows that pure WO3 photocatalysts had the one-dimensional nano-needle shape with a width of 80–100 nm and a length of 1.5–2 μm as presented in Fig. 3d. Figure 3b reveals that pure In2O3 photocatalysts were monodisperse nanoparticles with the size of approximately 50 nm as displayed in Fig. 3e. As for In/W-3 composite photocatalysts, the WO3 nano-needle photocatalysts were coated with a large number of In2O3 nanoparticles as shown in Fig. 3c. The corresponding EDS spectrum in Fig. S1 also demonstrated the coexistence of W, In, and O elements in the composite photocatalysts. The construction of heterostructure nano-photocatalysts was further confirmed by the HRTEM images. Figure 3d showed that pristine WO3 photocatalysts had typical nano-needle morphology with good crystallinity and the fringe spacing of 0.374 nm was assigned to the (020) crystal plane of WO3 [37]. The inset in Fig. 3e exhibited that pristine In2O3 was also crystalline and the fringe spacing of 0.291 nm was corresponding to (222) crystal plans of In2O3 [38]. It can be seen from Fig. 3f that In2O3 nanoparticles and WO3 nano-needles closely contacted with each other. Three different lattice planes were observed near the interface area. The fringe spacing of 0.199 nm, 0.291 nm, and 0.374 nm can be indexed to the (431), (222) crystal plans of In2O3 and the (020) crystal plane of WO3, respectively. The tightly contact interfaces between In2O3 and WO3 can facilitate the rapid separation and transfer of photo-generated electron–hole pairs, thus improving the photocatalytic activity of composite photocatalysts.

SEM images of a WO3, b In2O3, and c In/W-3, HRTEM images of d WO3, e In2O3, and f In/W-3
XPS analysis
The surface chemical compositions and elemental chemical states of In2O3, WO3, and In/W-3 were analyzed by XPS measurement. The full-scan XPS spectrum in Fig. 4a clearly demonstrated the presence of In, W, and O elements in In/W-3, indicating the coexistence of In2O3 and WO3 in the composite photocatalysts. The high-resolution XPS spectra of W, In, and O are shown in Fig. 4b–d. The W 4f XPS of pure WO3 in Fig. 4b showed two well-resolved peaks at 37.15 eV and 35.05 eV, corresponding to W 4f 5/2 and W 4f 7/2 two characteristic peaks of W6+, respectively [39]. Compared to pure WO3, the two characteristic peaks of W 4f in In/W-3 were shifted toward lower binding energies due to the interaction between WO3 and In2O3 photocatalysts. In Fig. 4c, the In 3d spectrum of In2O3 displayed two peaks at 450.95 eV and 443.40 eV, corresponding to In 3d 3/2 and In 3d 5/2 peaks of In3+ [40], while their positions in the In/W-3 made a significant shift toward higher values at 451.40 eV and 443.80 eV, respectively. It was noteworthy that the O1 s spectra in Fig. 4d were greatly different in WO3, In2O3, and In/W-3. The O 1s peak in In2O3 can be deconvoluted into two obvious bands at about 528.86 and 530.29 eV, which were assigned to the oxygen bond of In-O-In and the oxygen defects (Ov), respectively [41]. As for WO3, the O 1 s signal peak centered at about 529.80 eV can be ascribed to the crystal lattice oxygen in W–O bond. In terms of In/W-3 composite photocatalyst, the O 1 s spectrum can be deconvoluted into three obvious bands at about 529.16 eV, 529.71 eV, and 530.94 eV, which was similar to a combination of WO3 and In2O3. The above XPS results further confirmed the coexistence of In2O3 and WO3 in the In/W-3 heterostructure photocatalysts.

XPS spectra of WO3, In2O3 and In/W-3 photocatalysts: a full survey, b W 4f, c In 3d and d O 1s core levels
UV–Vis DRS
The UV–Vis DRS spectra of the WO3, In2O3, and In/W-x composite photocatalysts are plotted in Fig. 5a. Pure WO3 and In2O3 displayed relatively weak absorption intensities in visible light region and had absorption edges at 430 nm and 420 nm, respectively, which corresponded to a bandgap energy of 2.70 eV and 2.80 eV. Compared with the pure WO3 and In2O3 nano-photocatalysts, the In/W-x composite photocatalysts demonstrated stronger absorption in both UV and visible light regions, which may be ascribed to the interaction between WO3 and In2O3 nano-photocatalysts. At the same time, the absorption edges exhibited an obvious red shift phenomenon with the increasing amount of In2O3 in composite photocatalysts. When the usage of In2O3 was over 15%, the absorption in visible light region did not exhibit obvious enhancement. The above results demonstrated the construction of Z-scheme photocatalysts could effectively improve their utilization for sunlight and produced more photo-generated electrons and holes for photocatalytic reaction accordingly. Moreover, the band gap energies (Eg) of WO3, In2O3, and In/W-x composite photocatalysts can be estimated according to the modified Kubelka–Munk function (2):

a UV–Vis diffuse reflectance spectra of In2O3, WO3, and In/W-x photocatalysts, b Tauc plots of In2O3, WO3, and In/W-x photocatalysts
where a is the absorption coefficient, v is the light frequency, Eg is the band gap (eV), and A is a constant. From Tauc plots of different photocatalysts in Fig. 5b, Eg values of pure WO3 and In2O3 can be calculated to be approximately 2.79 and 2.83 eV, while Eg values of In/W-1, In/W-2, and In/W-3 are estimated to be 2.70, 2.43, and 2.41 eV, respectively. Remarkably, the introduction of In2O3 nanoparticles on WO3 nano-needle photocatalysts had obvious effects on narrowing band gap energies of composite photocatalysts, thereby promoting the absorption of visible light.
PL emission spectra and electrochemical properties
Photoluminescence spectroscopy was applied to study the separation processes of photo-generated electrons and holes in the as-prepared photocatalysts as shown in Fig. 6. Generally, a stronger fluorescence intensity stands for a higher recombination ratio of photo-generated electron–hole pairs [42]. Pure WO3 and In2O3 photocatalysts both exhibited strong PL peaks at the range of 340 nm to 450 nm under an excitation wavelength of 310 nm, indicating their high recombination rate of photo-induced carriers. After loading In2O3 nanoparticles on WO3, the PL intensities of heterogeneous In/W-x photocatalysts were remarkably lower than those of pure In2O3 and WO3. The PL spectra results indicated that the construction of Z-scheme heterostructure effectively inhibited the recombination of photo-generated electron–hole pairs.

Photoluminescence (PL) emission spectra of In2O3, WO3, and In/W-x with the excitation wavelength at 310 nm
The photoelectrochemical tests were also performed to study the interfacial charge transfer and separation process of the as-prepared photocatalysts. The transient photocurrent responses of pure WO3 and In/W-x heterogeneous photocatalysts were recorded under visible-light irradiation for four light on–off cycles. As shown in Fig. 7a, pure WO3 exhibited the lowest photocurrent response. The photocurrent intensities of In/W-x photocatalysts were obviously higher and increased gradually with the increasing contents of In2O3. These results demonstrated that the introduction of In2O3 nanoparticles improved separation efficiency of the photo-generated charge carriers in In/W-x photocatalysts. The charge separation efficiency of photocatalyst samples was further investigated by electrochemical impedance spectroscopy (EIS). Figure 7b displays the Nyquist plots of the photocatalyst photoelectrodes under visible-light irradiation. Normally, the charge carrier transfer process in photocatalysts can be reflected by the semicircle part of Nyquist curve at the high-frequency region, and the small radius results from efficient charge transfer [43]. Obviously, In/W-x displayed relatively smaller arc radius than pure WO3, indicating their faster interfacial electron transfer. The findings of transient photocurrent response and ESI measurement agreed well with the PL analysis. These above results demonstrated that Z-scheme heterostructure can significantly accelerate the separation and transfer of photo-generated electrons and holes in In/W-x composite photocatalysts, leading to higher photocatalytic activity.

a Transient photocurrent response curves and b electrochemical impedance spectra of In2O3, WO3, and In/W-x photocatalysts
Photocatalytic performance
The photocatalytic performances of the as-prepared photocatalysts were evaluated by decomposing MB, RhB, and TC under visible-light irradiation. Before irradiation, the dark adsorption experiments of organic dyes on photocatalysts were conducted and the concentration changes were shown in Fig. S2. It can be seen that for both MB and RhB dyes the adsorption–desorption equilibrium could be reached after 30 min. The photolysis results of two organic dyes in Fig. 8a and (d) displayed that the dyes had relatively high photochemical stability and their self-degradation could be neglected. At the presence of pure WO3 and In2O3 photocatalysts, only approximately 41.4% (39.6%) and 29.4% (30.8%) of MB (RhB) were decomposed after visible-light irradiation for 90 (120) min. Compared to single photocatalyst, the In/W-x binary Z-scheme photocatalysts exhibited substantially enhanced photocatalytic activities. The degradation rates of organic dyes increased with the increasing content of In2O3 in In/W-x. The In/W-1, In/W-2, and In/W-3 can remove 75.1% (54.4%), 89.7% (88.0%), and 92.4% (90.5%) of MB (RhB) dyes after visible-light irradiation for 90 (120) minutes, respectively. Meanwhile, the photocatalytic activity of In/W-3 was compared with that of TiO2-P25 in Fig. S3. Obviously, the photocatalytic degradation rate of organic dye over In/W-3 was much higher than TiO2-P25. The improved photocatalytic activity can be attributed to the construction of Z-scheme heterojunction between WO3 and In2O3, which led to wider visible-light absorption range and accelerated separation rate of photo-generated electron–hole pairs. The appropriate dosage of In2O3 in the composite photocatalysts was crucial for the enhancement of photocatalytic activity. When the content of In2O3 exceeded 10%, the In/W photocatalysts did not show significant increase. This could be explained as that too much In2O3 nanoparticle may interfere with light absorption and reduce the light intensity arriving at the surface of In/W nanocomposites [44].

Photocatalytic activities of different photocatalysts for the degradation of a MB and d RhB under visible-light irradiation, kinetic fit for the degradation of b MB and e RhB, kinetic rate constant for photocatalytic degradation of c MB and f RhB, g photocatalytic removal efficiency of TC by different photocatalysts
As shown in Fig. 8b, e, the photocatalytic degradation process of MB and RhB can be described by a pseudo-first-order kinetics reaction and expressed as Eq. (3):
where k is the kinetic rate constant (min−1). C0 and Ct are the concentrations of organic dyes at reaction time 0 and t, respectively. The corresponding reaction rate constants for different samples are displayed in Fig. 8c, f. The kinetic rate constants of MB removed by In/W-1, In/W-2, and In/W-3 photocatalysts were 0.016, 0.026, and 0.029 min−1, respectively, which were 1.59, 3.24, 3.70 times higher than that of WO3. Similarly, the degradation process of RhB by In/W-1, In/W-2, and In/W-3 photocatalysts also possessed much higher rate constants than WO3, which was consistent with the tendency of their degradation rate.
Besides these organic dyes, the colorless antibiotic TC was also used to evaluate the photocatalytic performance of the prepared photocatalysts. As shown in Fig. 9, the In/W-x exhibited higher photocatalytic activity for TC degradation than pure In2O3 and WO3. In/W-3 still had the best photocatalytic ability and could remove approximately 82.4% of TC molecules. The above results indicated that In/W Z-scheme nanocomposites can be utilized as an efficient photocatalysts for the degradation of organic pollutants.

Photocatalytic removal efficiency of TC by different photocatalysts
To study the effects of photocatalyst dosage on the photocatalytic degradation of organic dye, In/W-3 photocatalysts at different concentrations (0.4, 0.6, 0.8, 1.0, and 1.2 g/L) were used in the photocatalytic experiments and the results are presented in Fig. 10a. It is observed that the degradation efficiencies of both MB and RhB increased with the increasing concentration of photocatalysts from 0.4 g/L to 1.0 g/L. When the dosage of photocatalysts exceeded 1.0 g/L, photocatalytic degradation efficiency showed a slight decline. The phenomenon is due to that more photocatalysts can produce more active species to degrade organic dyes. But too much photocatalyst powder could result in a turbid solution, less visible light penetration, which was not conducive to the formation of photo-generated hole-electron pairs [45].

Effects of a photocatalyst dose and b pH on photocatalytic degradation of dyes using In/W-3 photocatalyst, the TOC removal efficiency of c MB and d RhB dyes using different photocatalysts, photocatalytic activities of In/W-3 photocatalyst after three cycles for the degradation of e MB and f RhB under visible-light irradiation
The pH value of dye solutions also obviously affected their photocatalytic degradation efficiency. As shown in Fig. 10b, under neutral conditions In/W-3 exhibited the highest photocatalytic activity for the degradation of both MB and RhB dyes. The degradation efficiencies of two organic dyes decreased significantly under acidic and alkaline pH conditions. The results can be explained as follows. The surface of In/W-3 photocatalysts could be easily protonated under acidic condition [46], which may reduce the electrostatic attraction between photocatalysts and cationic dyes (MB and RhB). Under alkaline conditions, the strong electrostatic interaction between photocatalysts and organic dyes can hinder the absorption of incident light.
Furthermore, the mineralization rates of organic dyes over WO3, In2O3, and In/W-x photocatalysts under visible-light irradiation were further evaluated by the total organic carbon (TOC) analysis. It can be found from Fig. 10c, d that In/W-x exhibited much higher TOC mineralization rate than pure WO3 and In2O3 photocatalysts. In/W-3 achieved the highest TOC removal efficiency (82.1% for MB, 72.5% for RhB) after 90-min light irradiation, indicating that the majority of organic dyes could be mineralized during the photocatalytic degradation process. The TOC results are also consistent with the decolorization results in Fig. 8a, d. It indicates that In/W-x photocatalysts have strong mineralization ability for MB and RhB organic dyes in the photocatalytic process.
In order to investigate the stability of the In/W nanocomposite photocatalysts, the recycling degradation experiments were carried out at neutral pH using 1.0 g/L of In/W-3 photocatalysts. As presented in Fig. 10e–f, after three cycles In/W-3 photocatalysts still remained relatively high photocatalytic performance toward the degradation of two organic dyes. The degradation rates of MB and RhB remained 86.6% and 86.4% at the third cycle at the presence of In/W-3. The used In/W-3 photocatalysts after three cycles were also characterized by XRD, BET, and SEM analysis. Figure S4(a) shows the XRD patterns of In/W-3 photocatalysts before and after photocatalytic degradation process. It can be seen that the location and intensity of characteristic peaks did not show significant changes after the photocatalytic reaction. The BET results in Fig. S4(b) indicated that the used In/W-3 photocatalyst had the similar N2 adsorption–desorption isotherms to that of the original sample. The BET specific surface areas of the samples before and after photocatalytic reaction were 28.79 m2/g and 27.14 m2/g. From the SEM images in Fig. S4(c-d), the morphologies of IN/W-3 photocatalysts had no obvious changes after three photocatalytic cycles. The above results demonstrated In/W-3 photocatalysts had outstanding structure stability under the visible-light irradiation.
Photocatalytic mechanism
The reactive oxygen species generated in the photocatalysis process were verified by the electron spin resonance (ESR) spectrum. As shown in Fig. 11a, b, the characteristic signal peaks of the DMPO-·OH and DMPO-·O2− adducts were detected under visible-light illumination. In/W-3 photocatalysts displayed much higher DMPO-·OH and DMPO-·O2− signal peaks than pure WO3 and In2O3 because of their relatively low electron–hole recombination rate. No obvious DMPO-·OH signals can be observed in pure In2O3 sample due to its less positive VB potential than H2O/·OH as presented in Fig. 13c. Meanwhile, the characteristic peaks of DMPO-·O2− cannot be found in pure WO3 sample owing to its more positive CB potential. The ESR results revealed that the In/W-3 photocatalysts can produce more ·OH and ·O2− radicals under visible-light irradiation.

ESR spectra of a DMPO-·OH in aqueous dispersion and b DMPO-·O2− in methanol dispersion, c photocatalytic degradation of MB by WO3, In2O3, and In/W-3 in the presence of t-BuOH, AO and BQ under visible-light irradiation
To further investigate the dominant species in photocatalytic degradation reaction of organic dyes, free radicals trapping experiments were conducted in the presence of three different scavengers: t-BuOH for ·OH, AO for photo-generated holes (h+) and BQ for ·O2− [47]. The effects of different scavengers on the photocatalytic activities of WO3, In2O3, and In/W-3 are depicted in Fig. 11c. T-BuOH and BQ had almost no effects on the photocatalytic reaction of WO3 compared with the photo-degradation result without scavengers, while AO resulted in a significantly declined photocatalytic efficiency of MB. Due to more positive CB potential (0.74 eV) of WO3 than the redox potential (− 0.33 eV) of O2/·O2−, no ·O2− could be produced in the WO3 photocatalytic system. Therefore, it can be speculated that h+ was the main active species in WO3 photocatalysts for photo-degradation of MB. Unlike WO3, in the presence of t-BuOH, In2O3 exhibited the similar photocatalytic activity to that without any scavengers. The photocatalytic degradation efficiency of MB decreased greatly after adding BQ scavenger, but improved slightly after adding AO scavenger due to the reduced recombination of photo-induced electron–hole pairs. The results indicated that ·O2− played a major role in In2O3 photocatalytic system. With regard to In/W-3, both AO and BQ adversely affected the photo-degradation efficiency of MB, suggesting the construction of Z-scheme between In2O3 and WO3. It was clear that h+ and ·O2−, rather than ·OH, were the main active species for In/W-3 during photocatalytic degradation process.
The degradation intermediates of two organic dyes were detected by LC–MS analysis [48]; the chemical structures and m/z values of degradation products are listed in Table 1 and Table 2. The possible photocatalytic degradation pathways of MB and RhB dyes by In/W-3 photocatalysts are proposed in Fig. 12. As shown in Fig. 12a, the MB molecules were mainly attacked by h+ and ·O2−, thereby leading to demethylation, deamination, oxidation, and a series of ring-opening reactions. They were gradually decomposed into several intermediates with aldehyde, carbonyl, and carboxyl groups, such as degradation products B (m/z = 224), C (m/z = 183), D (m/z = 141), E (m/z = 143), F(m/z = 161), G(m/z = 161), H(m/z = 118). Finally, these intermediates were mineralized to CO2 and H2O. Figure 12b displays the possible degradation pathway of RhB dye. Firstly, the N-deethylation occurred on the nitrogen atoms of RhB molecules due to the attacks of h+ and ·O2−, resulting in various intermediates including I (m/z = 359), J (m/z = 415), and K (m/z = 387). Then, the chromophore structures of these deethylated products were destroyed through cleavage of the conjugated xanthene structure, leading to the intermediates with low molecular weights including products L (m/z = 300), M (m/z = 239), N (m/z = 226), O (m/z = 195), and P (m/z = 217). The benzene ring structures of above intermediates were attacked continuously and oxidized to products Q (m/z = 114) and R (m/z = 118). Ultimately, the formed products could be mineralized to CO2 and H2O.

Photocatalytic degradation pathways of organic dyes: a MB, b RhB
Based on the above experimental results and band gap structures, the probable photocatalytic mechanism of Z-scheme In/W nanocomposite photocatalysts was proposed. As shown in Fig. 13a, WO3 has a band gap energy of 2.70 eV with the VB potential of 3.44 eV and the CB potential of 0.74 eV. In2O3 has a band gap energy of 2.80 eV with the VB potential of 2.17 eV and the CB potential of − 0.63 eV. Both WO3 and In2O3 belong to n-type semiconductors. According to previous reports, the Fermi level of n-type semiconductor is usually 0.20 eV lower than the CB potential [49]. Apparently, WO3 has lower Fermi level and larger work function (W2 = 6.23 eV) than In2O3 photocatalysts (W1 = 4.00 eV) [50, 51]. When the two photocatalysts contact with each other, the electrons will spontaneously flow from In2O3 to WO3 until their Fermi levels are aligned. Thus, In2O3 is positive charged due to the loss of electrons, and WO3 is negatively charged due to the accumulation of electrons at the interface. Then, an internal electric field is built at the In2O3/WO3 interface. At the same time, the energy band edges of In2O3 bend upward toward the interface and those of WO3 bend downward due to the transfer of electrons [52]. Figure 13c shows the migration process of photon-generated charge carriers between WO3 and In2O3 under visible-light irradiation. Firstly, WO3 and In2O3 can absorb visible light to generate the electron–hole pairs, respectively. Secondly, under the comprehensive effects of internal electric field, and band edge bending, the photo-generated electrons in the CB of WO3 can quickly migrate to the VB of In2O3 and recombine with the photo-generated holes. Simultaneously, the internal recombination of photo-induced charge carriers was effectively inhibited for both WO3 and In2O3 photocatalysts. As a result, for the Z-scheme heterogeneous In/W photocatalyst, the photo-generated electrons will accumulate in the CB of In2O3 and holes will gather in the VB of WO3. Finally, the leaving electrons in In2O3 can react with O2 to form ·O2− due to its more negative CB potential than the redox potential of O2/·O2−. The organic dyes were degraded by ·O2− to CO2 and H2O. The photo-generated h+ with strong oxidation ability in the VB of WO3 can directly take part in the photocatalytic degradation of organic dyes. In a word, the construction of Z-scheme structure between WO3 and In2O3 can not only promote the migration and separation of photo-generated carriers, but also retain the active species with high redox capacity for degradation of organic pollutants.

Schematic illustration of the possible charge transfer mechanism of photo-induced electrons and holes for the degradation of organic dyes: a before contact, b after contact, c after light irradiation
Conclusions
In summary, we successfully fabricated a novel all-solid state Z-scheme In2O3/WO3 heterogeneous photocatalyst through a facile two-step hydrothermal–solvothermal method. The obtained composite photocatalysts exhibited much higher photocatalytic degradation ability toward organic dyes and antibiotics under visible-light irradiation than pure WO3 and In2O3 semiconductor materials. When the content of In2O3 reached 15%, the corresponding photocatalyst In/W-3 achieved the highest dye degradation rate constants, which was 3.70 times higher than that of pure WO3. The enhanced photocatalytic performance can be attributed to the construction of Z-scheme heterojunction between WO3 nano-needles and In2O3 nanoparticles. The built-in electric field in this heterostructure accelerated the recombination of useless photo-generated holes and electrons, holding the photo-generated charge carriers with strong redox capacity for photocatalytic reaction. The free radical trapping experiments indicated that photo-excited holes and ·O2− in the Z-scheme photocatalytic system were the predominant active species for the organic dye degradation. The intermediates of two organic dyes were determined by HPLC–MS. This work provides new insights into the design of efficient heterostructure photocatalysts for the purification of textile dyeing wastewater.
References
Turcanu A, Bechtold T (2017) Cathodic decolourisation of reactive dyes in model effluents released from textile dyeing. J Clean Prod 142:1397–1405
Wang JD, Yao JC, Wang L et al (2020) Multivariate optimization of the pulse electrochemical oxidation for treating recalcitrant dye wastewater. Sep Purif Technol 230:115851
Ulson de Souza SMAG, Forgiarini E, Ulson de Souza AA (2007) Toxicity of textile dyes and their degradation by the enzyme horseradish peroxidase (HRP). J Hazard Mater 147:1073–1078
Xu R, Su M, Liu Y et al (2020) Comparative study on the removal of different-type organic pollutants on hierarchical tetragonal bismutite microspheres: adsorption, degradation and mechanism. J Clean Prod 242:118366
Anwer H, Mahmood A, Lee J et al (2019) Photocatalysts for degradation of dyes in industrial effluents: opportunities and challenges. Nano Res 12:955–972
Zhao Y, Wang R, Fang K et al (2019) Investigating the synergetic dispersing effect of hydrolyzed biomacromolecule cellulase and SDS on CuPc pigment. Colloids Surf B 184:110568
Wang H, Kong H, Zheng J et al (2020) Systematically exploring molecular aggregation and its impact on surface tension and viscosity in high concentration solutions. Molecules 25:1588
Reddy IN, Reddy CV, Shim J et al (2020) Excellent visible-light driven photocatalyst of (Al, Ni) co-doped ZnO structures for organic dye degradation. Catal Today 340:277–285
Nguyen CH, Lien TM, Van TTT et al (2020) Enhanced removal of various dyes from aqueous solutions by UV and simulated solar photocatalysis over TiO2/ZnO/rGO composites. Sep Purif Technol 232:115962
Fakhri H, Mahjoub AR, Cheshme Khavar AH (2016) Improvement of visible light photocatalytic activity over graphene oxide/CuInS2/ZnO nanocomposite synthesized by hydrothermal method. Mater Sci Semicond Process 41:38–44
Chen WC, Huang GY, Li XM et al (2019) Revealing the position effect of an alkylthio side chain in phenyl-substituted benzodithiophene-based donor polymers on the photovoltaic performance of non-Fullerene organic solar cells. ACS Appl Mater Interfaces 11:33173–33178
Santhosh C, Velmurugan V, Jacob G et al (2016) Role of nanomaterials in water treatment applications: a review. Chem Eng J 306:1116–1137
Fakhri H, Bagheri H (2020) Highly efficient Zr-MOF@WO3/graphene oxide photocatalyst: synthesis, characterization and photodegradation of tetracycline and malathion. Mater Sci Semicond Process 107:104815
Mardiroosi A, Mahjoub AR, Fakhri H (2017) Efficient visible light photocatalytic activity based on magnetic graphene oxide decorated ZnO/NiO. J Mater Sci: Mater Electron 28:11722–11732
Low J, Yu J, Jaroniec M et al (2017) Heterojunction photocatalysts. Adv Mater 29:1601694
Wang HL, Zang LS, Chen ZG et al (2014) Semiconductor heterojunction photocatalysts: design, construction, and photocatalytic performances. Chem Soc Rev 43:5234–5244
You JH, Guo YZ, Guo R et al (2019) A review of visible light-active photocatalysts for water disinfection: features and prospects. Chem Eng J 373:624–641
Deng F, Lu XY, Luo YB et al (2019) Novel visible-light-driven direct Z-scheme CdS/CuInS2 nanoplates for excellent photocatalytic degradation performance and highly-efficient Cr(VI) reduction. Chem Eng J 361:1451–1461
Zhao W, Feng Y, Huang HB et al (2019) A novel Z-scheme Ag3VO4/BiVO4 heterojunction photocatalyst: study on the excellent photocatalytic performance and photocatalytic mechanism. Appl Catal B Environ 245:448–458
Wang M, Tan GQ, Zhang D et al (2019) Defect-mediated Z-scheme BiO2−x/Bi2O2.75 photocatalyst for full spectrum solar-driven organic dyes degradation. Appl Catal B Environ 254:98–112
Xie RY, Zhang LP, Xu H et al (2015) Fabrication of Z-scheme photocatalyst Ag–AgBr@Bi20TiO32 and its visible-light photocatalytic activity for the degradation of isoproturon herbicide. J Mol Catal A: Chem 406:194–203
Chen SF, Hu YF, Meng SG et al (2014) Study on the separation mechanisms of photogenerated electrons and holes for composite photocatalysts g–C3N4–WO3. Appl Catal B Environ 150–151:564–573
Lu BA, Li XD, Wang TH et al (2013) WO3 nanoparticles decorated on both sidewalls of highly porous TiO2 nanotubes to improve UV and visible-light photocatalysis. J Mater Chem A 1:3900–3906
Bhosale NY, Mali SS, Hong CK et al (2017) Hydrothermal synthesis of WO3 nanoflowers on etched ITO and their electrochromic properties. Electrochim Acta 246:1112–1120
Ghosh S, Saha M, Paul S et al (2015) Maximizing the photo catalytic and photo response properties of multimodal plasmonic Ag/WO3-x heterostructure nanorods by variation of the Ag size. Nanoscale 7:18284–18298
Tie LN, Yu CF, Zhao YL et al (2018) Fabrication of WO3 nanorods on reduced graphene oxide sheets with augmented visible light photocatalytic activity for efficient mineralization of dye. J Alloys Compd 769:83–91
Zerjav G, Arshad MS, Djinovic P et al (2017) Electron trapping energy states of TiO2–WO3 composites and their influence on photocatalytic degradation of bisphenol A. Appl Catal B Environ 209:273–284
Chen JY, Xiao XY, Wang Y et al (2019) Fabrication of hierarchical sheet-on-sheet WO3/g-C3N4 composites with enhanced photocatalytic activity. J Alloys Compd 777:325–334
Liu XW, Xu JJ, Ni ZY et al (2019) Adsorption and visible-light-driven photocatalytic properties of Ag3PO4/WO3 composites: a discussion of the mechanism. Chem Eng J 356:22–33
Luo J, Zhou XS, Ma L et al (2017) Fabrication of WO3/Ag2CrO4 composites with enhanced visible-light photodegradation towards methyl orange. Adv Powder Technol 28:1018–1027
Xing YL, Que WX, Yin XT et al (2016) In2O3/Bi2Sn2O7 heterostructured nanoparticles with enhanced photocatalytic activity. Appl Surf Sci 387:36–44
Li B, Li XW, Shao CL et al (2019) Hierarchically porous In2O3/In2S3 heterostructures as micronano photocatalytic reactors prepared by a novel polymer-assisted sol-gel freeze-drying method. Ind Eng Chem Res 58:14106–14114
Li CH, Ming T, Wang JX et al (2014) Ultrasonic aerosol spray-assisted preparation of TiO2/In2O3 composite for visible-light-driven photocatalysis. J Catal 310:84–90
Ma DD, Shi JW, Zou YJ et al (2018) Multiple carrier-transfer pathways in a flower-like In2S3/CdIn2S4/In2O3 ternary heterostructure for enhanced photocatalytic hydrogen production. Nanoscale 10:7860–7870
An GW, Mahadik MA, Piao G et al (2019) Hierarchical TiO2@In2O3 heteroarchitecture photoanodes: mechanistic study on interfacial charge carrier dynamics through water splitting and organic decomposition. Appl Surf Sci 480:1–12
Chen XX, Li R, Pan XY et al (2017) Fabrication of In2O3–Ag–Ag3PO4 composites with Z-scheme configuration for photocatalytic ethylene degradation under visible light irradiation. Chem Eng J 320:644–652
Guo H, Jiang N, Wang HJ et al (2019) Pulsed discharge plasma assisted with graphene-WO3 nanocomposites for synergistic degradation of antibiotic enrofloxacin in water. Chem Eng J 372:226–240
Zhang F, Li XY, Zhao QD et al (2016) Rational design of ZnFe2O4/In2O3 nanoheterostructures: efficient photocatalyst for gaseous 1,2-dichlorobenzene degradation and mechanistic insight. ACS Sustainable Chem Eng 4:4554–4562
Guo H, Jiang N, Wang HJ et al (2019) Degradation of antibiotic chloramphenicol in water by pulsed discharge plasma combined with TiO2/WO3 composites: mechanism and degradation pathway. J Hazard Mater 371:666–676
Yang HR, Tian J, Bo YY et al (2017) Visible photocatalytic and photoelectrochemical activities of TiO2 nanobelts modified by In2O3 nanoparticles. J Colloid Interface Sci 487:258–265
Lei FC, Sun YF, Liu KT et al (2014) Oxygen vacancies confined in ultrathin indium oxide porous sheets for promoted visible-light water splitting. J Am Chem Soc 136:6826–6829
Chang F, Zhang X, Chen H et al (2019) Ag/AgCl nanoparticles decorated 2D-Bi12O17Cl2 plasmonic composites prepared without exotic chlorine ions with enhanced photocatalytic performance. Mol Catal 477:110538
Chen F, Yang Q, Wang SN et al (2017) Graphene oxide and carbon nitride nanosheets co-modified silver chromate nanoparticles with enhanced visible-light photoactivity and anti-photocorrosion properties towards multiple refractory pollutants degradation. Appl Catal B Environ 209:493–505
Chu XF, Hu T, Gao F et al (2015) Gas sensing properties of graphene-WO3 composites prepared by hydrothermal method. Mater Sci Eng, B 193:97–104
Ebrahimi R, Maleki A, Zandsalimi Y et al (2019) Photocatalytic degradation of organic dyes using WO3-doped ZnO nanoparticles fixed on a glass surface in aqueous solution. J Ind Eng Chem 73:297–305
Zhang GP, Chen DY, Li NJ et al (2019) Fabrication of Bi2MoO6/ZnO hierarchical heterostructures with enhanced visible-light photocatalytic activity. Appl Catal B Environ 250:313–324
Xie RY, Zhang LP, Xu H et al (2017) Construction of up-converting fluorescent carbon quantum dots/Bi20TiO32 composites with enhanced photocatalytic properties under visible light. Chem Eng J 310:79–90
Fakhri H, Mahjoub AR, Aghayan H (2017) Effective removal of methylene blue and cerium by a novel pair set of heteropoly acids based functionalized graphene oxide: adsorption and photocatalytic study. Chem Eng Res Des 120:303–315
Zheng RJ, Zhang M, Sun X et al (2019) Perylene-3,4,9,10-tetracarboxylic acid accelerated light-driven water oxidation on ultrathin indium oxide porous sheets. Appl Catal B Environ 254:667–676
Chen Y, Huang W, Sangwan VK et al (2019) Polymer doping enables a two-dimensional electron gas for high-performance homojunction oxide thin-film transistors. Adv Mater 31:1805082
Fu JW, Xu QL, Low JX et al (2019) Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst. Appl Catal B Environ 243:556–565
Zhang Z, Yates JT (2012) Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces. Chem Rev 112:5520–5551
Acknowledgements
We gratefully acknowledge the assistance of Professor Hua Zhou in improving the English language. This study was funded by (1) China National Key Technology R&D Program (grant number 2017YFB0309800); (2) Shandong Province Key Technology R&D Program (grant number 2017CXGC1006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xie, R., Fang, K., Liu, Y. et al. Z-scheme In2O3/WO3 heterogeneous photocatalysts with enhanced visible-light-driven photocatalytic activity toward degradation of organic dyes. J Mater Sci 55, 11919–11937 (2020). https://doi.org/10.1007/s10853-020-04863-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-04863-5