
Abstract
A new carbon precursor, soybean oil, was applied to the synthesis of ordered mesoporous carbon (SOMC) using an inverse replication method in conjunction with ordered mesoporous SiO2 molecular sieves (SBA-15) as a hard template. The effects of different carbonization temperatures (600, 700, and 800 °C) on the microstructure, degree of order of the pore structure, specific surface area, and pore size of the SOMC were investigated. Scanning electron microscopy showed that the SOMC was composed of rods approximately 1 μm in length and 0.5 μm in diameter. Transmission electron microscopy indicated that carbonization temperatures in the range of 600–800 °C produced, the SOMC samples having a suitably ordered mesoporous structure. N2 adsorption–desorption analysis confirmed that the Brunauer–Emmett–Teller specific surface area of the SOMC was 882 m2/g with a pore diameter of 3.5 nm after processing at 600 °C.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
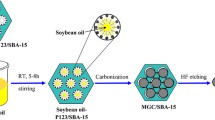
Ordered mesoporous carbon (OMC) consists of both ordered and regular mesoporous channels and also exhibits chemical inertness, electrical conductivity, and high mechanical strength [1]. Consequently, this material is widely used in catalysis, adsorption, separation, and other applications [2]. The synthesis of OMC generally uses carbon precursors arranged regularly as a result of the action of a template, after which these are carbonized to form OMC [3]. The templating agents can be divided into hard and soft templates [4, 5]. The hard template methods employ solid materials, such as ordered mesoporous silica, having a fixed and ordered structure. As shown in Scheme 1, the carbon precursor fills the hard template channels based on capillary action and is subsequently carbonized. After the removal of the hard template, the OMC reproduced the structure of the hard template, but in reverse [4, 6].

A schematic of the synthesis of ordered mesoporous carbon by the hard template method
Wang et al. [7] synthesized OMC specimens having Brunauer–Emmett–Teller (BET) specific surface areas in the range of 640–1100 m2/g and pore sizes of 3.4–7.7 nm using SBA-15 (an ordered mesoporous silica molecular sieve) as a hard template, sucrose as a carbon source, and boric acid as a pore-expanding agent. Wang et al. [8] also employed SBA-15 as a hard template together with functionalized ionic liquid as the carbon and nitrogen source to prepare nitrogen-doped OMC. The specific surface area and pore size of the resulting material were 446.3 m2/g and 3.7 nm, respectively. Fang et al. [9] synthesized OMC with a BET specific surface area of 889 m2/g and a 3.9 nm pore size using finger citron essential oil as the carbon source and SBA-15 as the hard template. Lee et al. [10] employed AlCl3 to treat hexagonal mesoporous silica (HMS) aluminosilicate to obtain the hard template Al-HMS. Formaldehyde and phenol were loaded into the pores of this template and polymerized into a phenolic resin. After the Al-HMS was carbonized and removed, the mesoporous carbon SUN-2 was obtained, with a specific surface area of 1056 m2/g and pore sizes of 0.6 and 2 nm.
Soft-templating methods typically involve the self-assembly of a carbon precursor under the guidance of a surfactant to form an OMC [5]. In 2004, Dai et al. [5] first applied the soft template technique to the preparation of ordered mesoporous carbon films. A mixture of 4-vinyl pyridine and resorcinol was used as the carbon precursor to form ordered and well-oriented nanostructures through strong hydrogen bonding between the pyridine groups and resorcinol [11]. Tanaka et al. [1] synthesized the ordered mesoporous carbon film material COU-1 with a BET surface area of 1274 m2/g and pore diameter of 6.2 nm through self-assembly using pyrocatechol, formaldehyde, and triethyl acetate as the carbon precursors and non-ionic F127 as a surfactant. Ghimbeu et al. [12] synthesized OMC based on solvent volatilization induced self-assembly, with phloroglucinol/glyoxylic acid as the carbon precursor and a structure-directing agent.
The hard template method can be used to synthesize mesostructures that cannot be obtained by organic surfactant self-assembly techniques. Additionally, hard templates show stable physicochemical properties, and so a variety of chemical and heat treatments can be used to prepare highly crystallized OMC products [13].
Vegetable oils are an environmentally friendly and renewable resource primarily composed of C, H, O, the main components of which are triglycerides [14]. An example is glyceryl trilinoleate, which has the molecular formula C57H98O6, a carbon ratio of approximately 0.55 (C/(H + O)) and a sucrose content of 0.36. For these reasons, vegetable oils are extremely useful as raw materials for sources of carbon materials. Studies have also been conducted on the use of vegetable oils as raw feedstocks for the preparation of carbon materials, such as carbon nanotubes. Raziah et al. [15] applied a mixture of castor oil and ferrocene as the raw material in conjunction with thermal decomposition in the range of 300–400 °C using microwave radiation under N2 atmosphere to prepare tubular carbon nanotubes with diameters of 7–50 nm. Paul et al. [16] synthesized multi-walled carbon nanotubes with diameters of 80–100 nm via a chemical vapor deposition technique using coconut oil as the carbon source, iron as the catalyst, and N2 as a carrier for the vaporized precursor. Suriani et al. [17] employed palm oil as a carbon source to synthesize multi-walled and single-walled carbon nanotubes with length of approximately 110 µm by chemical vapor deposition.
In the present study, SBA-15 was used as a hard template together with soybean oil as the carbon precursor to synthesize SOMC samples at 600, 700, or 800 °C. The morphologies and microstructures of these materials were characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), N2 adsorption–desorption (to determine Brunauer–Emmett–Teller (BET) specific surface areas), X-ray diffraction (XRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS).
Materials and methods
Materials
P123 (a PEO-PPO-PEO triblock copolymers), with an average Mn of approximately 5800, was procured from Sigma-Aldrich (USA). Tetraethyl orthosilicate (TEOS, 98%), hydrofluoric acid (40%), and soybean oil (reagent grade)were purchased from Aladdin (China); H2SO4 (98%) and hydrochloric acid (36%) were purchased from the Sinopharm Chemical Reagent Co. Ltd. (China), and ethanol (95%) was purchased from the Yantai Shuangshuang Chemical Co. Ltd (China).
Experimental
Synthesis of SBA-15
The synthesis of SBA-15 has been previously reported by Zhao et al. [18]. In this process, 4 g P123, 120 g hydrochloric acid (2 mol/L), and 30 g deionized water were combined and stirred for 0.5 h to obtain a transparent solution, after which, 8.5 g TEOS was added with stirring and the mixture was maintained at 40 °C for 24 h. The mixture was subsequently transferred to a hydrothermal reactor and crystallized at 100 °C for 72 h. After filtering and drying (at 80 °C for 12 h), the resulting white powder was calcined at 550 °C (applying a ramp rate of 1 °C/min) for 6 h in air to obtain the SBA-15.
Synthesis of SOMC
The synthesis of SOMC was performed based on various techniques previously reported in the literature [19, 20]. In the present work, 5 mL anhydrous ethanol, 76 μL concentrated sulfuric acid (98%), and 1 g SBA-15 were mixed with ultrasonication for 10 min. Following this, 1.25 g soybean oil was added to the mixture with 10 min of continued ultrasonication. Thereafter, the mixture was maintained at 100 °C for 6 h and then 160 °C for another 6 h. The resulting solid was calcined at 600 °C (at a ramp rate of 5 °C/min) under N2 for 6 h to obtain the carbon–silica composite, termed C/SiO2-600. This material was subsequently soaked in 10% hydrofluoric acid for 24 h, then filtered off and washed with deionized water until the wash water was neutral to produce the OMC-600. Samples treated at 700 and 800 °C, are referred to herein as SOMC-700 and SOMC-800, respectively.
Characterization methods
TEM images of the samples were acquired using a Tecnai G2 F20 (FEI, USA), while the morphologies and microstructures of these materials were assessed by SEM with an EVO18 instrument (Zeiss, Germany). N2 adsorption–desorption data were obtained using an ASAP 2020 surface area and porosity analyzer (Micromeritics, USA). XRD patterns were acquired with a SmartLab X-ray diffractometer (Rigaku, Japan) with Cu Kα radiation, operating at 40 kV and 30 mA. Raman spectra were acquired measurement using an Evolution Raman spectrometer (Horiba, France) at a laser wavelength of 633 nm. XPS was performed with an Escalab 250 Xi spectrometer (Thermo Scientific, USA) using Al Kα radiation. Thermogravimetric analysis (TGA) was conducted using a STA449F5 synchronous thermal analyzer (Netzsch, Germany) at a heating rate of 10 °C/min under N2 or air.
Results and discussion
TEM and SEM
Figure 1a–c presents TEM images of the SOMC specimens obtained using the different calcination temperatures. Looking along the direction perpendicular to the pore channels, an ordered strip-shaped pore structure is evident. Conversely, the SOMC-600 exhibits a regular 2D hexagonal pore structure in the direction parallel to the pore channels. The silica walls of the SBA-15 were found to contain micropores or to have a submesoporous structure. In the nano-pouring process, the soybean oil entered the micropores and submesoporous structure and carbonized to form carbon nanowires connected to one another via carbon rods. After the SBA-15 was removed, the result was a 2D hexagonal ordered mesoporous structure [21, 22]. Wang et al. [23] synthesized OMC at 850 °C under Ar using Al-SBA-15 as the template and furfuryl alcohol as the carbon precursor, and produced TEM images showing a similar highly ordered mesoporous structure. Figure 1d–f presents TEM images of the C/SiO2 composites produced at different calcination temperatures. The carbon is seen to be evenly dispersed in the SBA-15 mesopores to form a composite nanostructure [24]. Compared to the SOMC products, the C/SiO2 composites also exhibit a more obvious ordered structure. Figure 2 provides SEM images of the OMC specimens and C/SiO2 composites produced at the various calcination temperatures. These materials were composed of rods approximately 1 μm in length and 0.5 μm in diameter and had morphologies very similar to those of the OMC and SBA-15 previously synthesized by Ignat et al. [25].

TEM images of SOMC and C/SiO2 composite samples

SEM images of SOMC and C/SiO2 composite samples
N2 adsorption–desorption
The nitrogen adsorption–desorption isotherms of the SOMC samples synthesized at the various calcination temperatures are shown in Fig. 3a. These isotherms are all type IV and exhibit type H4 hysteresis loops, indicating that these were mesoporous materials. These type H4 hysteresis loops can be attributed to the presence of compound pores in the OMC, as are apparent in Fig. 3b. This figure demonstrates that the pore diameter distributions of the OMC specimens were bimodal with diameters in the range of 2.5–5.0 nm. In our previous work [26], an OMC prepared using SBA-15 as a hard template and sucrose as the carbon source also generated a type IV isotherm with a type H4 hysteresis loop. Additionally, Fuertes [27] synthesized an OMC with SBA-15 as the hard template and furfuryl alcohol as the carbon source and this material produced a typical IV isotherm with a type H1 hysteresis loop. The isotherms obtained from the SOMC materials were not closed but rather each contained a long, narrow gap over the relative pressure range from approximately 0 to 1. This result can possibly be ascribed to the expansion of the soybean oil at high temperatures [28]. The average pore diameter of each OMC was in the range of 3.5–3.9 nm (Table 1). Chang et al. [29] employed SBA-15 as a hard template and furfuryl alcohol as the carbon source to prepare an OMC with a pore diameter of 3.7 nm via heating at 800 °C under N2. The BET surface areas of the present SOMC specimens were in the range of 783–882 m2/g (Table 1), and the SOMC-600 had the highest area of 882 m2/g with a slight decrease in area on changing the carbonization temperature from 600 to 800 °C. Ding et al. [30] used SBA-15 as a hard template and d-fructose as the carbon source to produce an OMC with a BET specific surface area of 351 m2/g and a pore size of 3.2 nm at 900 °C, while Wang et al. [31] applied SBA-15 as a hard template and sucrose as the carbon source to synthesize an OMC with a BET specific surface areas of 620 m2/g and pore diameter of 4.6 nm at 900 °C under argon. The isotherms obtained from C/SiO2 specimens produced at different calcination temperatures are presented in Fig. 3c. Each isotherm can be classified as type IV, indicating that these materials were mesoporous, and the isotherms all demonstrate the same multilayer adsorption. During the capillary condensation stage associated with relative pressure values of 0.4–0.8, the adsorption and desorption lines were separated, and wide hysteresis loops were generated similar to H1 loops. Compared to the SOMC, the C/SiO2 isotherms were closer to that of SBA-15 [32]. Gokulakrishnan et al. [32] synthesized SBA-15 using P123 as a structure-directing agent and tetraethyl orthosilicate as a silica source, and observed type IV sorption isotherms with H1 hysteresis loops. Babić et al. [33] produced SBA-15/carbon cryogel composites via the solgel polycondensation of resorcinol and formaldehyde followed by freeze-drying and subsequent pyrolysis. These materials were found to give type IV isotherms with hysteresis loops that indicated mesoporous structures. However, an SBA-15/carbon composite made using sucrose as the carbon source generated a type I isotherm suggesting a microporous material. The pore diameter distributions of the C/SiO2 specimens synthesized at the different carbonization temperatures were found to be highly similar to one another (Fig. 3d). Each demonstrated a unimodal distribution with average pore diameters of approximately 5.5 nm (Table 1). The BET surface areas of these samples were in the range of 358–568 m2/g (Table 1), and so smaller than the values for the SOMC specimens. This difference is attributed to the silicon matrix occupying the pores in the former material. Wu et al. [3] synthesized a carbon–silicon composite with a surface area of 338 m2/g using 1, 4-bis (triethoxysilyl) benzene as the carbon precursor and P123 as a template agent, which had a mesoporous structure and an increased surface area of 1397 m2/g after removal of the silica. Babić et al. [34] prepared silica/cryogel carbon composites with specific surface areas of 350–520 m2/g using SBA-15 as a hard template and found specific surface areas of 533–604 m2/g after removing the silicon.

N2 adsorption/desorption isotherms and pore diameter distributions for a and b SOMC and c and d C/SiO2 composite specimens
XRD
Figure 4a provides the small-angle XRD patterns obtained from the various SOMC samples and demonstrates the presence of a broad diffraction peak in each at a 2θ of approximately 1.3°. This peak corresponds to the 100 crystal plane, indicating that the OMC samples all had an orderly hexagonal structure [35]. Yang et al. [26] synthesized OMC materials using SBA-15 as a hard template and sucrose as the carbon source and these specimens produced XRD patterns showing a wide diffraction peak at a 2θ of 1.1°, corresponding to the 100 crystal plane. Figure 4b shows the wide-angle XRD spectra of the SOMC samples. These patterns each contain two peaks at 24° and 43° that correspond to the 002 and 100 crystal planes, respectively, demonstrating the formation of semi-graphitized carbon [36]. With increases in the carbonization temperature, the diffraction peak representing the 002 crystal plane was not significantly enhanced, while that related to the 100 plane gradually increased in intensity. These results are similar to wide-angle XRD data reported for the material synthesized by Xie et al. [37]. Figure 4c presents the small-angle XRD patterns of the C/SiO2 composites generated at different calcination temperatures. Three diffraction peaks are evident at 2θ of 0.8°, 1.4° and 1.7°, which corresponded to the 100, 110, and 200 crystal planes. These patterns are thus in agreement with the pattern obtained for SBA-15 by Hartmann et al. [35] and suggest that these composites had a 2D, six-sided channel structure. The 2θ values for the 100 crystal planes of the C/SiO2 composites were relatively constant despite variations in the calcination temperature, confirming that the pore diameters in these materials were not affected by temperature [4, 38]. This result is consistent with the pore diameter distribution data reported by Hartmann. Figure 4d shows the wide-angle XRD patterns of these same C/SiO2 composites. An intense peak is apparent at a 2θ value of approximately 22° together with a less intense peak at 44°, corresponding to the 002 and 100 crystal planes of carbon, respectively. These results also demonstrate the presence of semi-graphitized carbon [36].

XRD patterns of SOMC and C/SiO2 composite specimens: a, c small-angle patterns and b, d wide-angle patterns
Raman spectroscopy
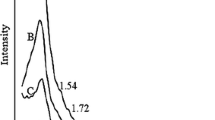
Figure 5 provides the Raman spectra of the various SOMC specimens. Each spectrum contains two peaks at 1580 and 1334 cm−1 that corresponds to graphite (G) and disorder (D) bands, respectively. The G-band indicates that a hexagonal graphite lattice was formed, while the D-band shows imperfect hexagonal symmetry within the graphite structure [39]. With increases in the carbonation temperature, the ID/IG ratios (0.99, 1.04 and 0.98) did not change significantly. Atchudan et al. [40] prepared graphitic mesoporous carbon at different temperatures and the resulting Raman spectra had ID/IG ratios of 0.93 (for the material prepared at 800 °C), 0.79 (900 °C) and 0.98 (1000 °C). Xie et al. [37] synthesized C/SiO2 composites (containing 5.51 wt% carbon) at different carbonization temperatures (600, 700, 800, 900, and 1000 °C), and the Raman spectra of these specimens exhibited increases in the ID/IG ratio with increasing temperature. The ID/IG value is proportional to the number of defect sites in the carbon material, which in turn plays a role in the performance of the OMC as a catalyst support. Li et al. [41] combined iron phthalocyanine (FePc) with reduced graphene oxide (RGO), mesoporous carbon vesicles (MCVs), and OMC to synthesize a series of electrochemical catalysts and found that the FePc/OMC combination promoted the oxygen reduction reaction to the greatest extent. Raman analyses showed that the ID/IG ratio of the OMC (1.93) which was higher than those for the RGO and MCVs. These data suggested that the OMC had more edge plane-like defect sites that promoted electron transfer reactions and acting as adsorption sites for catalysis. Hou et al. [42] coated OMC-x materials (where x is the sucrose-to-silica mass ratio, with values of 0.6, 1.25 or 3) onto glassy carbon (GC) electrodes for the electrocatalytic oxidation of hydroquinone and found that the OMC-0.6/GC electrode exhibited the strongest electrocatalytic response. The ID/IG ratios of these materials indicated that the OMC-0.6 surface contained the highest concentration of edge plane-like defect sites. When OMC is used as a catalyst support, there is a clear correlation between the ID/IG ratio and catalytic performance. However, when OMC is employed as an adsorbent, the correlation between the ID/IG ratio and absorption performance is weaker. Tang et al. [43] prepared an OMC with embedded Co nanoparticles as an adsorbent for rhodamine B removal. The IG/ID ratio of the Co/OMC was found to be higher than that of the raw OMC, and so the degree of graphitization of the pore wall structure in the Co/OMC was evidently increased. Tang proposed that this high level of graphitization improved the acid/base and oxidation resistance of the material. Armandi et al. [44] applied SBA-15 and MCM-48 as hard templates together with sucrose as the carbon precursor to synthesize C-15N2 and C-48, respectively, to assess applications in hydrogen storage. The Raman data showed that the ID/IG ratios of the C-15N2 and C-48 were similar, although the C-15N2 demonstrated a greater hydrogen uptake ongoing from 0 to 650 mmHg pressure.

Raman spectra of SOMC specimens
XPS
Figure 6a provides the C1s XPS spectra of the SOMC samples prepared at the different carbonization temperatures. The characteristic peak observed in each spectrum in the range of 284.80–284.82 eV corresponds to C=C bonds and accounts for 78.87% (600 °C), 70.21% (700 °C) and 73.29% (800 °C) of the total integrated intensity. With increases in the carbonization temperature, the intensity of this peak is seen to have first decreased and then increased slightly. The peak at 286.29–286.32 eV is attributed to C–O bonds, and accounts for 9.44% (600 °C), 14.73% (700 °C) and 11.48% (800 °C) of the total integrated intensity. The peak at 289.20–289.50 eV results from O–C=O groups [45], and accounts for 11.69% (600 °C), 15.06% (700 °C) and 15.23% (800 °C). It is evident that the XPS and Raman data are in agreement. In the case of the OMC-700, the ID/IG ratio was 1.04, indicating that this specimen had the highest proportion of disordered carbon. Correspondingly, the XPS results demonstrate that the total integrated intensity of the peaks due to C–O and O–C=O groups were highest in the OMC-700 pattern. These results suggest that the presence of disordered carbon may be related to the oxidation of carbon. Figure 6b shows the O1s XPS spectra obtained from the OMC materials. The peak at 531.50–531.96 eV corresponds to C=O bonds and accounts for 24.85% (600 °C), 18.81% (700 °C), and 27.95% (800 °C) of the total integrated intensity. The peak at 532.94–533.06 eV is attributed to C–O bonds and accounts for 75.15% (600 °C), 81.19% (700 °C), and 72.05% (800 °C) of the total integrated intensity [46].

XPS spectra obtained from the SOMC specimens
TGA
Figure 7 shows the TGA curves acquired from the SOMC materials either under nitrogen or air. Under nitrogen and over the range of 50–550 °C, the data show mass increases of 1.47% (SOMC-600), 1.71% (SOMC-700), and 2.13% (SOMC-800), possibly caused by the adsorption of nitrogen. As the temperature was further increased from 550 to 800 °C, the masses returned to the original values, indicating that the adsorbed nitrogen was desorbed. There was thus essentially no mass loss over the range of 50–800 °C. In contrast, under air, the SOMC samples showed no mass loss over the range of 50–500 °C but a sharp drop in mass between 500 and 600 °C. The mass losses were 94.66% (SOMC-600), 99.23% (SOMC-700) and 94.90% (SOMC-800). These results are ascribed to the oxidation of these materials under the air atmosphere.

TGA curves obtained from the SOMC specimens
Conclusions
A series of SOMC specimens were prepared using soybean oil as the carbon source and SBA-15 as a hard template at carbonization temperatures of 600, 700, and 800 °C under a nitrogen atmosphere. TEM images showed that these SOMC materials maintained an ordered mesoporous structure. The BET specific surface areas of the samples were in the range of 783–882 m2/g and the average pore diameters were in the range of 3.5–3.9 nm. Raman spectra indicated the coexistence of a hexagonal graphite lattice (producing a G-band) and imperfect hexagonal symmetry within the graphite structure (producing a D-band). TGA results demonstrated minimal mass loss over the temperature range of 50–800 °C under nitrogen but losses as high as 94.66–99.23% under air.
References
Tanaka S, Nishiyama N, Egashira Y, Ueyama K (2005) Synthesis of ordered mesoporous carbons with channel structure from an organic–organic nanocomposite. Chem Commun 16:2125–2127
Ma TY, Liu L, Yuan ZY (2013) Direct synthesis of ordered mesoporous carbons. Chem Soc Rev 42(9):3977–4003
Wu Z, Pang J, Lu Y (2009) Synthesis of highly-ordered mesoporous carbon/silica nanocomposites and derivative hierarchically mesoporous carbon from a phenyl-bridged organosiloxane. Nanoscale 1(2):245–249
Jun S, Joo SH, Ryoo R, Kruk M, Jaroniec M, Liu Z, Terasaki O (2000) Synthesis of new, nanoporous carbon with hexagonally ordered mesostructure. J Am Chem Soc 122(43):10712–10713
Liang C, Hong K, Guiochon GA, Mays JW, Dai S (2004) Synthesis of a large-scale highly ordered porous carbon film by self-assembly of block copolymers. Angew 116(43):5909–5913
Wan Y, Shi Y, Zhao D (2008) Supramolecular aggregates as templates: ordered mesoporous polymers and carbons†. Chem Mater 20(3):932–945
Wang G, Zhang Z, Wang J, Li N, Hao Z (2015) Study of the influence of pore width on the disposal of benzene employing tunable OMCs. Ind Eng Chem Res 54(3):1074–1080
Wang R, Yu Y, Zhang Y, Lv H, Chen A (2017) Synthesis of nitrogen-doped mesoporous carbon for the catalytic oxidation of ethylbenzene. IOP Conf Ser Mater Sci Eng 207:012012
Fang Y, Dai W, Chen L, Ma N (2016) Facile synthesis of ordered mesoporous carbon with finger citron residue as carbon precursor. Mater Lett 174:246–248
Lee J, Yoon S, Oh SM, Shin CH, Hyeon T (2000) Development of a new mesoporous carbon using an HMS aluminosilicate template. Adv Mater 12(5):359–362
Kim SH, Misner MJ, Xu T, Kimura M, Russell TP (2004) Highly oriented and ordered arrays from block copolymers via solvent evaporation. Adv Mater 16(3):226–231
MateiGhimbeu C, Vidal L, Delmotte L, Le Meins JM, VixGuterl C (2014) Catalyst-free soft-template synthesis of ordered mesoporous carbon tailored using phloroglucinol/glyoxylic acid environmentally friendly precursors. Green Chem 16(6):3079
Zhao DY, Wan Y, Zhou WZ (2013) Ordered mesoporous molecular sieve materials. Higher Education press, Beijing
Lee DW, Park YM, Lee KY (2009) Heterogeneous base catalysts for transesterification in biodiesel synthesis. Catal Surv Asia 13(2):63–77
Raziah AZ, Junizah AR, Saifuddin N (2012) Synthesis of carbon nanotubes using natural carbon precursor: castor oil. AIP Conf Proc 1482:564–567
Paul S, Samdarshi SK (2011) A green precursor for carbon nanotube synthesis. New Carbon Mater 26(2):85–88
Suriani AB, Azira AA, Nik SF, Md Nor R, Rusop M (2009) Synthesis of vertically aligned carbon nanotubes using natural palm oil as carbon precursor. Mater Lett 63(30):2704–2706
Zhao D (1998) Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 279(5350):548–552
Yang M, Gao Q (2011) Copper oxide and ordered mesoporous carbon composite with high performance using as anode material for lithium-ion battery. Microporous Mesoporous Mater 143(1):230–235
Barrera D, Dávila M, Cornette V, de Oliveira JCA, López RH, Sapag K (2013) Pore size distribution of ordered nanostructured carbon CMK-3 by means of experimental techniques and Monte Carlo simulations. Microporous Mesoporous Mater 180:71–78
Rumplecker A, Kleitz F, Salabas EL, Schüth F (2007) Hard templating pathways for the synthesis of nanostructured porous Co3O4. Chem Mater 19(3):485–496
Fan J, Wang T, Yu C, Tu B, Jiang Z, Zhao D (2004) Ordered, nanostructured tin-based oxides/carbon composite as the negative-electrode material for lithium-ion batteries. Adv Mater 16(16):1432–1436
Wang X, Tian Y (2010) Templated synthesis of ordered mesoporous carbons with tailored structures and morphologies. Chin J Chem 28(5):858–862
Ren Y, Yang L, Wang L, Xing H, Wu H (2015) Microwave absorption properties of Fe-doped ordered mesoporous carbon (CMK-3)/silica matrix nanocomposites with magnetic multi-resonance mechanisms. Nano 10(08):1550110
Ignat M, Van Oers CJ, Vernimmen J, Mertens M, Potgieter-Vermaak S, Meynen V, Cool P (2010) Textural property tuning of ordered mesoporous carbon obtained by glycerol conversion using SBA-15 silica as template. Carbon 48(5):1609–1618
Yang L, Yuan H, Wang S (2019) Preparation and application of ordered mesoporous carbon-based solid acid catalysts for transesterification and epoxidation. J Porous Mater 26:1435–1445
Fuertes AB (2004) Synthesis of ordered nanoporous carbons of tunable mesopore size by templating SBA–15 silica materials. Microporous Mesoporous Mater 67(2–3):273–281
Demir M, Kahveci Z, Aksoy B, Palapati NKR, Subramanian A, Cullinan HT, Gupta RB (2015) Graphitic biocarbon from metal-catalyzed hydrothermal carbonization of lignin. Ind Eng Chem Res 54(43):10731–10739
Chang B, Fu J, Tian Y, Dong X (2013) Multifunctionalized ordered mesoporous carbon as an efficient and stable solid acid catalyst for biodiesel preparation. J Phys Chem Lett 117(12):6252–6258
Ding S, Zheng S, Xie M, Peng L, Guo X, Ding W (2011) One-pot synthesis of boron-doped mesoporous carbon with boric acid as a multifunction reagent. Microporous Mesoporous Mater 142(2–3):609–613
Wang DW, Li F, Chen ZG, Lu GQ, Cheng HM (2008) Synthesis and electrochemical property of boron-doped mesoporous carbon in supercapacitor. Chem Mater 20(22):7195–7200
Gokulakrishnan N, Kania N, Léger B, Lancelot C, Grosso D, Monflier E, Ponchel A (2011) An ordered hydrophobic P6mm mesoporous carbon with graphitic pore walls and its application in aqueous catalysis. Carbon 49(4):1290–1298
Babić B, Kokunešoski M, Miljković M, Prekajski M, Matović B, Gulicovski J, Bučevac D (2012) Synthesis and characterization of the SBA–15/carbon cryogel nanocomposites. Ceram Int 38(6):4875–4883
Babić B, Kokunešoski M, Miljković M, Matović B, Gulicovski J, Stojmenović M, Bučevac D (2013) New mesoporous carbon materials synthesized by a templating procedure. Ceram Int 39(4):4035–4043
Hartmann M, Vinu A (2002) Mechanical stability and porosity analysis of large-pore SBA-15 mesoporous molecular sieves by mercury porosimetry and organics adsorption. Langmuir 18(21):8010–8016
Wang T, Zhang C, Sun X, Guo Y, Guo H, Tang J, He J (2012) Synthesis of ordered mesoporous boron-containing carbon films and their corrosion behavior in simulated proton exchange membrane fuel cells environment. J Power Sources 212:1–12
Xie P, Wang Z, Zhang Z, Fan R, Cheng C, Liu H, Guo Z (2018) Silica microsphere templated self-assembly of a three-dimensional carbon network with stable radio-frequency negative permittivity and low dielectric loss. J Mater Chem C 6(19):5239–5249
Wang L, Dong X, Jiang H, Li G, Zhang M (2014) Ordered mesoporous carbon supported ferric sulfate: a novel catalyst for the esterification of free fatty acids in waste cooking oil. Fuel Process Technol 128:10–16
Wu M, Ai P, Tan M, Jiang B, Li Y, Zheng J, He X (2014) Synthesis of starch-derived mesoporous carbon for electric double layer capacitor. Chem Eng J 245:166–172
Atchudan R, Perumal S, Karthikeyan D, Pandurangan A, Lee YR (2015) Synthesis and characterization of graphitic mesoporous carbon using metal-metal oxide by chemical vapor deposition method. Microporous Mesoporous Mater 215:123–132
Li M, Bo X, Zhang Y, Han C, Guo L (2014) Comparative study on the oxygen reduction reaction electrocatalytic activities of iron phthalocyanines supported on reduced grapheme oxide, mesoporous carbon vesicle and ordered mesoporous carbon. J Power Sources 264:114–122
Hou Y, Guo L, Wang G (2008) Synthesis and electrochemical performance of ordered mesoporous carbons with different pore characteristics for electrocatalytic oxidation of hydroquinone. J Electroanal Chem 617:211–217
Tang L, Cai Y, Yang G, Liu Y, Zeng G (2014) Cobalt nanoparticles-embedded magnetic ordered mesoporouscarbon for highly effective adsorption of rhodamine B. Appl Surf Sci 314:746–753
Armandi M, Bonelli B, Bottero I, Areán CO, Garrone E (2007) Synthesis and characterization of ordered porous carbons with potential applications as hydrogen storage media. Microporous Mesoporous Mater 103:150–157
Im H, Kim J (2012) Thermal conductivity of a graphene oxide-carbon nanotube hybrid/epoxy composite. Carbon 50(15):5429–5440
Zhao ZK, Dai YT, Lin JH, Wang GR (2014) Highly-ordered mesoporous carbon nitride with ultrahigh surface area and pore volume as a superior dehydrogenation catalyst. Chem Mater 26(10):3151–3161
Funding
This study was funded by the National Natural Science Foundation of China (21962001), China; the National Natural Science Foundation of China (21862001), China; Ningxia Scientific and Technological Innovation Leading Personnel Training (KJT2017006), China; The talent highland of research and development of high-value utilization technology of low-quality resources in Ningxia, China; and New Catalytic Process in Clean Energy Production (ZDZX201803), China, are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, W., Yuan, H. & Ke, Y. Preparation and characterization of ordered mesoporous carbon based on soybean oil. J Mater Sci 55, 6525–6536 (2020). https://doi.org/10.1007/s10853-020-04480-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-04480-2