
Abstract
Poly(vinyl alcohol-co-ethylene) (PVA-co-PE) nanofibers (flexible and effective bonding together) were combined with stiff and flame-resistant sepiolite nanorods (SEP) to fabricate composite nanofibrous aerogels (NFAs) through a versatile and gelation-free freeze-drying method. The PVA-co-PE nanofibers prepared based on a high-yield Melt-Extrusion-Phase-Separation process could form a continuous fibrous structure, and the fibrous networks and surface wettability of composite NFAs could be tuned by the amount of SEP used. The introduction of an appropriate amount of SEP improved mechanical properties of composite aerogels, while excessive SEP would lead to a decrease in compression strength of them. The mechanical properties of composite NFAs are shown to be governed by a combination of stiffness of SEP and effective bonding of PVA-co-PE nanofibers. More importantly, the presence of SEP can substantially improve flame resistance of aerogels. After a simple thermal chemical vapor deposition treatment with methyltrichlorosilane, the surface of composite NFAs become rougher and siloxane nanoparticles became visible. With this deposition, hydrophilic NFAs became hydrophobic as a significant surface property modification. Furthermore, the maximum stress at 60% strain went up from 15.57 to 19.17 kPa and the resilience of composite aerogels improved. Thermal properties results confirmed silane coating can improve the thermal stability and insulation characteristics of polymeric aerogel. The manufactured NFAs are demonstrated as good thermal insulators (0.0274 W m−1 K−1), excellent refractories, efficient water/oil separators (45–108 g/g, 78.58–91.76%) and reusable absorbers. Consequently, the multifunctional silane-coated PVA-co-PE/SEP composite NFAs could be envisaged for several demanding applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanofibrous aerogels (NFAs) are known to be the lightest solid material in the world with high porosity, which are fabricated through the assembly of various nanofibers, including PAN nanofibers [1], cellulose nanofibers [2, 3] and boehmite nanofibers [4]. Hence, they have received significant attention for various applications related to absorption [5], thermal insulation [6], flame retardance [7] and catalysis [8]. There are three main types of NFAs: inorganic NFAs, organic NFAs and composite NFAs. However, owing to the fragility of the inorganic nanofibers, the inorganic aerogels could generally suffer from high brittleness and low flexibility [9]. In comparison with inorganic nanofibers, organic nanofibers exhibit better flexibility, better damage resistance maintaining a and continuous organic fibrous structure that could effectively enhance the mechanical properties and stability of the resultant aerogels [10, 11]. However, the organic nanofibers normally are flammable and have low thermal stability, thus limiting their applications at high temperature.
In order to overcome these limitations, preparation of composite NFAs was thought to be an efficient method, by designing them to combine the beneficial properties of its components. Numerous investigations have shown that the incorporation of nanomaterials such as clay and inorganic nanofillers into organic NFAs with continuous fibrous structure could provide the aerogels with excellent mechanical properties, superior thermal properties and good flame resistance [12, 13]. The rigid nanofillers can support the structure of NFA and obviously improve the mechanical properties of it [1, 14]. Meanwhile, the introduction of nanofillers could effectively restrict the movement of organic polymer molecular chains to enhancing the thermal stability of aerogels [15] and reducing the radiative contribution to the thermal insulation retaining their mechanical properties at high temperatures [12]. Moreover, inclusions of clay and other nanofillers act as barriers which can substantially reduce the transfer of heat and prevent the flammable volatiles from getting into the organic fibrous structure during the process of combustion [16, 17].
Sepiolite nanorod (SEP) is one kind of fibrous natural nontoxic clay with high specific surface area, high porosity, remarkable chemical and mechanical stability and excellent flame resistance [18, 19], which has widely been used in various applications related to absorption [20], flame retardance [12], catalysis [21] and bioengineering [22, 23]. Furthermore, abundant hydroxyl groups on the surface of SEP could generate strong interactions between the SEP and matrix, which efficiently improved the adhesion between the matrix and SEP nanofillers [24, 25]. This behavior of SEP substantially enhanced the mechanical properties of the resulting composites and makes the fibrous SEP to be dispersed uniformly into a polymer matrix. The incorporation of SEP can significantly reduce the degradation rate of composites in high temperature applications, which might be associated with a preventative effect of SEP on the mobility of polymer molecular chains [26]. During combustion of the composite matrix, SEP can quickly form a dense silicate-rich layer on the surface of matrix, which effectively reduce heat or fire from damaging these composites [12]. SEP was usually chosen as a reinforced nanofiller to prepare two-dimensional composite materials based on PVA polymer [27,28,29]. In this paper, we introduce SEP into three-dimensional thermoplastic polymeric materials to fabricated PVA-co-PE/SEP composite NFAs and the effect of SEP on the properties of thermoplastic polymeric NFAs was investigated.
Herein, the thermoplastic polymeric nanofibers—PVA-co-PE nanofibers which forms the matrix of the composite fabricated using the routine mass production method of Melt-Extrusion-Phase-Separation, were chosen for the continuous fibrous structure. As in another earlier study, this polymer system is chosen as a suitable material for the preparation of NFAs [5] and fibrous SEP was introduced into aerogels to form composite NFAs. The cross-linking method is highly effective, which forms strong chemical covalent bonding between matrix nanofibers improving the mechanical properties of the composite NFAs [14, 30, 31]. Glutaraldehyde (GA) with two functionalities was selected as the cross-linker to bond the nanofibers tightly together [32, 33]. The amphiphilic PVA-co-PE/SEP composite NFAs transited to superhydrophobic and oleophilic after silanization via a simple thermal chemical vapor deposition (TCVD) method with methyltrichlorosilane. The siloxane nanoparticles were deposited onto both surface and inside of NFAs. The resulting silane-coated PVA-co-PE/SEP composite NFA could achieve excellent resilience and elastic durability, good thermal stability, low thermal conductivity (0.0274 W m−1 K−1), flame resistance, remarkable sorption ability (45–108 g/g, 78.58–91.76%) and sorption recyclability.
Materials and methods
Materials
PVA-co-PE resin (62 mol% vinyl alcohol) with a molecular weight of 7000 and cellulose acetate butyrate ester (CAB, Model 381-20) was supplied from Nippon Gohsei (Japan) and Eastman Chemical Company (United States), respectively. Acetone (AR), glutaraldehyde aqueous solution (AR, GA), tert-butanol (AR, t-BuOH) and acetic acid (AR, 99.5%) were obtained from Sinopharm Chemical Reagent Co., Ltd. Sepiolite nanorods (SEP, ~ 25 nm thick and ~ 1 μm long, Fig. S1), methyltrichlorosilane (AR) and most organic solvents were supplied by Sigma-Aldrich Co., Ltd (United States), including chloroform (AR), ethanol (AR), hexane (AR) and toluene (AR). Deionized water was used for all experiments. All chemical reagents were used as received.
Fabrication of PVA-co-PE nanofibers
The PVA-co-PE nanofibers were obtained through a mass production method called Melt-Extrusion-Phase-Separation, which has been elaborated in a previous procedure [34]. In brief, the PVA-co-PE resin and CAB powder were dried at 80 °C for 24 h in a vacuum drying oven, and then, the two polymers were mixed in a blend ratio of CAB/PVA-co-PE = 80/20. The blend was gravimetrically fed into twin-screw extruder (D = 16 mm, L/D = 40; Eurolab16, Thermo-Haake Co.). The feed rate was 6 g min−1, and the screw speed was 50 rpm. Barrel temperature profiles were 200, 205, 210, 215, 220 and 225 °C. The extrudates were hot-drawn at the die exit by a take-up device and water-cooled to room temperature. The CAB matrix was removed from extrudates via Soxhlet extraction of acetone to obtain PVA-co-PE nanofibers.
Preparation of composite suspensions
To synthesize composite suspensions, 0.7 g PVA-co-PE nanofibers was dispersed into 70 mL of mixture solvent (acetic acid solution (1 vol%) and t-BuOH at a mass ratio of 4:1) through homogenizing at speed of 12000 rpm for 20 min to obtain uniform PVA-co-PE nanofiber suspensions. 0.3 g of SEP was blended with 30 mL of acetic acid aqueous solution (1 vol%) at 12000 rpm to create SEP suspensions. Then, the prepared PVA-co-PE and SEP suspensions were mixed together to form 0.7 wt% PVA-co-PE and 0.3 wt% SEP composite suspensions. 1 mL of cross-linker (GA) was added to 0.7 wt% PVA-co-PE and 0.3 wt% SEP composite suspensions with mechanical mixing and forming the resultant composite nanofibrous suspensions. All these steps were conducted at room temperature.
Fabrication of composite NFAs
The newly prepared composite suspensions were frozen in a cryogenic refrigerator (− 65 °C) and subsequently freeze-dried in a lyophilizer for 48 h under vacuum (5–10 Pa) to gain the un-cross-linked composite nanofibrous aerogels (NFAs). Finally, the un-cross-linked composite NFAs were heated at 75 °C for 4 h in air to achieve cross-linked samples.
Fabrication of silane-coated composite NFAs
The superhydrophobic composite NFAs were obtained by employing a simple thermal chemical vapor deposition (TCVD) method. The PVA-co-PE/SEP composite samples and 5 mL of methyltrichlorosilane were placed in a sealed desiccator at 80 kPa below atmospheric pressure and heated to 50 °C for 12 h to make silanization of composite NFAs. The treated PVA-co-PE/SEP composite NFAs samples were continuously kept in a vacuum desiccator to remove the remaining methyltrichlorosilane and the by-product (HCl) [31].
Characterization and measurements
The densities of NFAs were determined by dividing their mass by their volume. The porous morphology and microstructure of NFAs were observed by a field emission scanning electron microscopy (FE-SEM, JSM-5800, JEOL, Japan). Attenuated total reflection-Fourier transform infrared spectrometry (ATR-FTIR, EQUINOX55, Bruker, Germany) was used to characterize any alterations of the NFAs before and after treatments. X-ray diffractometer (Dmax-1200, Rigaku, Japan) was carried out to analyze the phase composition of NFAs. The surface wettability of NFAs was measured by a contact angle meter (DSA100, Kruss, Germany) with water or oil volume of 3 μL at room temperature. The compression behaviors of NFAs were measured by an Instron 5969 (Instron, the USA) equipped with a 100 N load. Cylindrical NFA with a diameter of ∼ 30 mm was used, and the strain rate was set at 30 mm/min for the tests with 40, 60 and 80% compressive strain. The cycle loading–unloading cyclic test was performed at a compressive strain of 60% with a strain rate of 300 mm/min. The thermal stability of NFAs was characterized by a thermogravimetric analyzer (TG209F1, Netzsch, Germany). About 5 mg of samples was placed into quartz crucible and heated from ambient temperature to 700 °C at a heating rate of 10 °C/min under nitrogen environment (20 ml/min). The thermal conductivity of NFAs was measured by the transient hot-wire method (TC3000, Xiatech Electronics, China). Two NFAs with a thickness of 10 mm clamped the hot wire and tested with the voltage of 0.8 V at ambient temperature.
The porosity of NFAs
The porosity of NFAs was calculated by the following equation:
where η is the porosity, V0 is the volume of the NFAs, m is the mass of the solid constituents, and ρ is the density of solid constituents (include PVA-co-PE nanofibers, SEP and deposition).
The absorption capacity of oil and organic solvent
The silane-coated PVA-co-PE/SEP composite NFA (about 20 mg) was immersed into various types of pure oils and organic solvents. After the aerogel absorbed saturation, the sample was taken out from the pure liquid and excess liquid on the surface of it was removed by using a filter paper. The saturated silane-coated composite NFA was weighed immediately to avoid any evaporation. The absorption capacity was calculated by the following equation:
where \( W_{0} \) and \( W \) are the weight of the silane-coated composite NFA before and after absorption, respectively.
When the absorption volume is taken into account, the absorption capacity was then calculated as:
where \( \rho_{0} \) is the density of the original silane-coated composite NFAs, \( \rho \) is the density of the liquid.
Result and discussion
Silane-coated PVA-co-PE/SEP composite NFAs
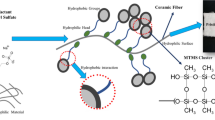
The fabrication process of silane-coated PVA-co-PE/SEP composite nanofibrous aerogels (NFAs) is demonstrated graphically in Fig. 1. The process was designed by a simple, environment-friendly and gelation-free freeze-drying method. The PVA-co-PE nanofibers obtained through Melt-Extrusion-Phase-Separation were selected as the skeletal materials to construct the aerogels, and glutaraldehyde (GA) was chosen as cross-linker to add into the aerogels to make their pores stiffer. The introduction of rigid nanofiller-SEP could enhance the structural stability of aerogels. The PVA-co-PE nanofibers and SEP were first homogenized in the solvent mixture to form uniform composite suspensions; the cross-linker was subsequently added into the composite suspensions. Then, the as-prepared composite suspensions were frozen and dried into un-cross-linked composite aerogels with good formability. The cross-linked PVA-co-PE/SEP composite NFAs with elastic resilience were obtained by heat treatment on un-cross-linked composite aerogels. After the heat treatment process, the binding between PVA-co-PE nanofibers from the weak physical bonds to strong chemical covalent bonds increases its strength and stability of the composite system. The stable SEP could prevent volume shrinkage of aerogels during cross-linking, and the density of samples obviously decreased from 12.31 to 11.42 mg cm−3. The silanization of composite NFAs occurred at 50 °C, and the color of composite NFAs changed from white to brown (Fig. S2). Moreover, the silane-coated PVA-co-PE/SEP exhibited superhydrophobicity and oleophilicity.

Schematic diagram of the fabrication process of silane-coated PVA-co-PE/SEP composite NFAs (ρ = 12.57 mg cm−3)
Morphology and structure of NFAs
As shown in Fig. 2a, d, all NFAs showed a continuous fibrous network structure, and their pores are highly interconnected. The PVA-co-PE nanofibers were deformed in both composite NFAs and silane-coated samples, which might be associated with the cross-linking between PVA-co-PE nanofibers. The nano-sized rods wound around the skeleton of aerogels tightly and guaranteed stable fibrous networks. The suspensions were frozen by cryogenic refrigerator (− 65 °C), and the freezing rate of composite suspensions was quite low, leading larger crystals. After sublimation under vacuum conditions, large breaks and poor integrity would potentially exist in the generated NFAs [35, 36]. Therefore, the introduction of t-BuOH with low surface tension (2.36 × 10−2 N m−1) could efficiently decrease the surface tension of composite suspensions and prevents large breaks during the freezing process [37]. Meanwhile, excessive content of t-BuOH leads to a decrease in center height of frozen samples [1]. 20% (v/v) was the optimal t-BuOH content, and the formed NFAs could achieve good formability [38].

FE-SEM of aerogels (ρ = 12.57 mg cm−3): a, b, c PVA-co-PE/SEP composite NFAs, d, e, f silane-coated PVA-co-PE/SEP composite NFAs
It is observed from Fig. 2b and e that the nano-sized SEP distributed uniformly in the pores of PVA-co-PE NFAs, which could tune the pore size and structure of aerogels. The SEP with a diameter of about 25 nm agglomerated slightly after the sublimation was observed, which was often found in compounds containing SEP (Fig. 2c, f). After silane coating, aerogels’ surfaces became rougher and nanoparticles became visible (Fig. 2f), which might be proposed to be the dehydrochlorination reaction between the active silicon-chlorine bonds on the methyltrichlorosilane and the residual hydroxyl groups on the PVA-co-PE nanofiber and SEP (Fig. S3). These siloxane nanoparticles could equip the composite samples with superhydrophobicity and oleophilicity.
Figure 3a shows the ATR-FTIR spectra of SEP, cross-linked PVA-co-PE NFAs, PVA-co-PE/SEP and silane-coated PVA-co-PE/SEP composite NFAs. After heat cross-linking treatment at 75 °C, the ether groups were formed by the reaction of hydroxyl and aldehyde groups. The absorption peak at 1137 cm−1 was corroborated the presence of ether groups, which was slightly overlapped with the absorption peak of C–C. The presence of ether groups confirmed successful cross-linking. As shown in the spectra of SEP, the characteristic peaks at 3562 and 3690 cm−1 were associated with stretching vibrations of hydroxyl groups in the external surface and octahedral Mg sheet, respectively, and the coordination bands at 1016 cm−1 assigned to stretching vibrations of Si–O in the Si–O-Si groups of the tetrahedral sheet [21, 39]. All of the characteristic peaks mentioned above were found at the spectra of composite NFAs, which demonstrated the successful introduction of SEP in composite aerogels. With increasing SEP contents in composite samples, the intensity of characteristic peaks of SEP increased. The well-disperse SEP in aerogels was certified by this phenomenon (Fig. 3b). However, once the SEP contents exceeded 30 wt%, with further increase in SEP content, the intensity of characteristic peaks remained nearly constant (Fig. 3c). This might be the result of agglomeration and inhomogeneous distribution of SEP in NFAs with high concentration. Therefore, the optimum content of SEP was found to be 30 wt%. The new characteristic absorption peaks at 781 and 1273 cm−1 corresponded to the asymmetric stretching vibrations of Si–O–Si and C-Si in C–Si–O units, respectively, at the spectra of silane-coated PVA-co-PE/SEP composite NFAs [31]. This might be proposed to be related to silanization of composite aerogels.

a ATR-FTIR spectra of SEP, cross-linked PVA-co-PE NFAs, PVA-co-PE/SEP composite NFAs and silane-coated PVA-co-PE/SEP composite NFAs, b, c ATR-FTIR spectra of composite NFAs with various SEP contents: b 5–30% SEP, c 30–50% SEP
The XRD patterns are illustrated in Fig. 4a, the primary peaks at 2θ = 7.18, 20.48, 26.54 and 34.94° corresponded to the characteristic diffraction peaks of SEP [21]. And the diffraction peaks were found at the XRD patterns of PVA-co-PE/SEP and silane-coated PVA-co-PE/SEP composite NFAs. With the increasing SEP concentrations in composite samples, the intensity of diffraction peaks of SEP increased significantly (Fig. 4b). The well-distributed SEP in aerogel was also demonstrated by XRD spectra. However, as shown in Fig. 4c, the variation of diffraction peaks intensity of SEP in composite NFAs is irregular, which could be ascribed to a massive aggregation of SEP with high content.

a XRD patterns of SEP, cross-linked PVA-co-PE NFAs, PVA-co-PE/SEP composite NFAs and silane-coated PVA-co-PE/SEP composite NFAs, b, c XRD patterns of composite NFAs with various SEP contents: b 5–30% SEP, c 30–50% SEP
Surface wettability of NFAs
Superhydrophobicity of the surface of composite NFAs is a crucial factor, which is beneficial to improve oil–water selectivity and separation efficiency of aerogels. With both hydrophilic hydroxyls and hydrophobic long alkyl chains, PVA-co-PE nanofibers exhibit amphiphilic, which allows PVA-co-PE nanofibers to take in both polar and nonpolar liquids. The PVA-co-PE nanofibers were assembled into aerogels through cross-linking method, and the water contact angle (WCA) of pure PVA-co-PE NFA surface is 144.4°, indicating the high hydrophobicity of pure cross-linked PVA-co-PE NFAs [5]. However, when the nanofillers (SEP) were introduced into aerogels, the WCA of the samples decreased substantially. As shown in Fig. 5b, the uncoated composite NFAs absorbed the water droplet of 3 μL immediately, and the WCA reduced to 0° in less than 1 s. This is proposed to be related to the hydrophilicity of SEP and well-distributed SEP in aerogel samples, as shown in Fig. 5a. With increasing SEP concentrations in aerogels, the WCA of composite aerogels decreased (shown in Fig. S4). Furthermore, the oil contact angle (OCA) of composite aerogels reduced to 0° within 1 s, which is proposed to the hydrophobic long alkyl chains in composite aerogels, allows uptake of oils (Fig. 5c). Therefore, the oil–water selectivity of composite NFAs was poor. After thermal chemical vapor deposition (TCVD), the samples allow modification to tune the amphiphilicity to greater hydrophobicity. It is obvious that the siloxane nanoparticles were bonded uniformly on the surface of nanofibers and SEP (Fig. 5d), and they could act as hydrophobic coating. As shown in Fig. 5e, the WCA of silane-coated composite NFAs was as high as 153.3° at 0 s, and a water droplet could maintain its original shape on the surface of silane-coated composite NFAs after 180 s. This could be explained by the consumption of surface hydroxyls through reacting with methyltrichlorosilane at 50 °C, and the formation of hydrophobic siloxane coating on the surface of NFAs. Moreover, the OCA of silane-coated composite NFAs demonstrated the oleophilicity of them (Fig. 5f). After the silane coating, the oil–water selectivity of composite aerogels improved substantially. Therefore, high efficiency of oil–water separation ensures their superior oil and organic solvent absorbance from water.

Surface wettability of the aerogels (ρ = 12.57 mg cm−3): FE-SEM of a PVA-co-PE/SEP composite NFAs, d silane-coated PVA-co-PE/SEP composite NFAs, b, c WCA and OCA measurement of PVA-co-PE/SEP composite NFAs, respectively, e, f WCA and OCA measurement of silane-coated PVA-co-PE/SEP composite NFAs, respectively
The resulting composite samples prepared by employing TCVD method show superhydrophobic and oleophilic for the whole aerogels and not only for the outmost surface. The gaseous methyltrichlorosilane could penetrate inside cellular fibrous structure of aerogels and deposit on the surface of exposed nanofibers. As shown in Fig. S5a, b, dyed water droplets bead up not only on the surface but also in the internal cross section of silane-coated composite NFAs, demonstrating that the siloxane coating make both the exterior and interior of PVA-co-PE/SEP composite NFAs hydrophobic. The superhydrophobic inner part of composite aerogels prevented water absorption of nanofibers, and the oleophilicity characteristic inside the composite NFAs could absorb more oil.
Mechanical properties of NFAs
The aerogels synthesized from flexible nanofibers showed poor resilience and durability after large strain cyclic compression [31, 40]. This was due to the low-stiffness nanofibers being unable to support the three-dimensional porous structure of aerogels under large compressive strains (> 60%) [41]. Therefore, by introducing stiffer SEP could make the compressibility of composite aerogels tunable while enhancing its properties. As shown in Fig. 6a, with increasing SEP concentrations, the stress of aerogels increased gradually. The rigid SEP not only acted as a support to prevent a collapse of aerogels but also obviously improved the strength of samples. However, when the SEP content exceeded 30 wt%, the strength of composite aerogels decreased (Fig. 6b). This might be proposed that the PVA-co-PE nanofibers content decreased with increasing SEP concentrations (almost the same density), and the effective bonding between PVA-co-PE nanofibers was reduced in composite NFAs. Therefore, the compression strength of composite NFAs decreased. As shown in Fig. 6c, 30 wt% was the optimal SEP content, and the mechanical stress of composite NFAs is 15.57 kPa. We also found that, after the silane coating, the compressive stress improved from 15.57 to 19.17 kPa. This is ascribed to that methyltrichlorosilane reacted with hydroxyls in PVA-co-PE nanofibers and SEP during the process of TCVD, where leads to higher strength of pore wall. The stress was 49.66 kPa at a strain of 80%, and the value was higher than most polymeric aerogel [30, 42].

Mechanical properties of silane-coated PVA-co-PE/SEP composite NFAs (ρ = 12.57 mg cm−3): a, b compressive cures of composite NFAs with various SEP contents: a 5–30% SEP, b 30–50% SEP, c compressive stress of samples with various SEP contents at 60% strain, d compressive stress–strain curves of silane-coated PVA-co-PE/SEP composite NFAs at different strains (ε = 40%, 60% and 80%), e cyclic stress–strain curves of silane-coated PVA-co-PE/SEP composite NFAs with ε of 60%, f the corresponding Young’s modulus and maximum stress as a function of the compressive test cycles
The silane-coated composite aerogels could sustain large deformations (more than 90%) without collapse, because of their high porosity (> 98%) and the strong combination between nanofibers, and they completely recover to their initial shape after release of the loading (Fig. 6d, Movie S1). Figure 6d displays a typical behavior of viscoelastic, energy-dissipative and highly deformable material [43]. Two distinct regions were observed in these curves, a linear elastic region at ε < 40%, which corresponded to blending of nanofibers and pores; and a densification region at ε > 40%, corresponding to the densification of pores [1]. The silane-coated samples could recover to their initial shape without fractures after being subjected to large deformation (ε > 80%). In contrast to pure cross-linked PVA-co-PE NFAs, the silane-coated composite NFAs showed higher compression modulus and better elastic resilience. The stress of composite NFAs was 19.17 kPa at a strain of 60%, much higher than that of pure PVA-co-PE NFAs (9.89 kPa at 60% strain) [5]. This significant improvement is the result of a well-distributed rigid SEP in the three-dimensional (3D) network nanofiber structure.
The silane-coated composite NFAs also displayed superior cycle performance under the large strain of 60%. The cyclic compression stress–strain curves of silane-coated composite NFAs at a strain of 60% are shown in Fig. 6e. The silane-coated composite NFAs almost could keep their original structure after the cyclic compression test. It is obvious to obverse the hysteresis loops and plastic deformation (ca. 12.30%) in the cures after 500 repeated compression, which is attributed to the energy-dissipation of aerogels during the cyclic compression [1]. Meanwhile, it could maintain over 86% of maximum stress after 500 cycles, as shown in Fig. 6f. The resilience and durability of silane-coated composite NFAs were greater than other hybrid polymeric aerogels [31].
Thermal properties of NFAs
The thermal stability of NFAs was investigated by thermogravimetric analysis (TGA). The TGA results of NFAs are shown in Fig. 7. Three main stages of degradation were observed in these TGA curves. For the cross-linked PVA-co-PE NFAs, the first weight loss stage, ranges from 286 to 332 °C, was caused by the degradation of vinyl alcohol. The second weight loss stage, from 332 to 407 °C, was attributed to degradation of ethylene component [44, 45]. The third weight loss stage occurred above about 417 °C, which was attributed to the degradation of ether groups. Almost no residue (1.67 wt%) was left after TGA test. The introduction of thermal stable SEP could improve significantly the thermal stability of composite NFAs. The initial decomposition temperature of composite NFA was high than that of cross-linked NFA, and about 25 wt% residue was found at the end of the cycle (as shown in Fig. 7). The rigid SEP acts as a barrier, which impedes the mobility of the flexible PVA-co-PE nanofibers in the process of heating and leading to more residues [15, 46].The zeolite water and the crystal water were decomposed at low temperatures, resulting in less than 30 wt% residue. For silane-coated samples, the thermal decomposition of the aerogel sample was retarded, and the decomposition temperature increased [31].

TGA cures of cross-linked PVA-co-PE NFAs, PVA-co-PE/SEP composite NFAs and silane-coated PVA-co-PE/SEP composite NFAs
Aerogel is an effective thermal insulator with low thermal conductivity due to its high porosity (> 98%) [47]. The thermal conductivity of aerogel samples with different compositions at room temperature is listed in Table 1. The cross-linked PVA-co-PE NFAs exhibited thermal conductivity of 0.0296 W m−1 K−1, which is lower than the previously reported aerogel insulators [12]. We attributed this result to the higher porosity of cross-linked PVA-co-PE NFA. When the SEP was added into NFAs, the thermal conductivity of them decreased to 0.0280 W m−1 K−1. This was associated with a number of factors: (1) the pores of PVA-co-PE NFA were filled with SEP and separated to smaller, the size of some pores in NFA was even smaller than the mean free path of air; [48] (2) SEP could act as phonon barriers and substantially reduce solid thermal conductivity [17]. (3) SEP with small diameters and large specific surface area could enhance the shielding ability and reduce the heat radiation of NFA [9]. As illustrated in Fig. S6, with increasing SEP concentrations in composite NFAs, the thermal conductivity of samples decreased gradually. After TCVD treatment, the aerogel exhibited the lowest thermal conductivity of 0.0274 W m−1 K−1, resulting from an insulation layer formed on the surface of NFAs, and thermal propagation was obstructed by the insulation coating. The thermal insulation of PVA-co-PE/SEP composite NFA was much better than those of commercially available insulators, such as polystyrene foam (0.04–0.06 W m−1 K−1) [49], even better than those of previously published aerogels (0.03–0.05 W m−1 K−1) [40, 50]. The silane-coated composite NFA could act as an excellent thermal insulator.
Flame resistance of NFAs
The PVA-co-PE nanofiber is a thermoplastic polymeric nanofiber with high flammability at normal circumstances. Therefore, the pure PVA-co-PE NFA inherits its flammability nature, which could be ignited easily once exposed to flame and continued to burn completely. This nature of PVA-co-PE NFA increases the risk of fire during the removal of flammable solvents. Motivated by this background in order to give flame-resistant to aerogels, the thermally stable and flame-retardant SEP was selected to improve the flame resistance of aerogel [12]. The combustion tests were performed, and the cylindrical NFAs with diameters of ~ 30 mm were directly subjected to fire. The pure PVA-co-PE NFA was burned up, and molten droplets could be found during the process of combustion (Fig. 8a, Movie S2), confirming the inflammability of pure PVA-co-PE aerogel. After incorporation of SEP in the aerogels, only the surface of aerogel was ignited. This could be proposed that the PVA-co-PE nanofibers on the surface of composite aerogel are flammable and the air filled in the pores of composite NFA acted as a combustion-supporter. However, the weak fire was extinguished in less than 10 s, and the black residue on the surface of composite NFA was observed (Fig. S7). This might be attributed to that SEP would form a protective silicate-rich surface layer and increase the rate of transformation of the carbon sources (PVA-co-PE) into char [12, 51]. Both of them could protect the interior of composite NFAs and prevent the flame and heat from entering composite NFA. Therefore, the internal structure of composite NFA was not destroyed, as shown in Fig. S8f, indicating that the existence of SEP could improve the flame-resistant properties of NFAs. The silane-coated PVA-co-PE/SEP composite NFAs also exhibited good flame resistance (Fig. 8b, Movie S3).

Combustion test of aerogels: a cross-linked PVA-co-PE NFA, b PVA-co-PE/SEP composite NFA
With the increasing SEP concentrations in aerogels, the flame resistance of composite NFAs increased. As illustrated in Fig. S8, the composite NFA with low SEP concentrations completely shrank and almost transformed into ashes after burning. The optimum content of SEP was 30%, and the internal structure and geometric volume of composite NFA remained nearly constant. If the content of SEP continued to increase, the PVA-co-PE/SEP composite NFA would suffer from high fragility with poor resilience [9]. Besides, excessive SEP content formed a massive agglomeration of SEP in the composite aerogel, resulting in inhomogeneous composite NFA.
Oil and organic solvent absorbency of NFAs
These silane-coated composite NFAs also showed excellent sorption selectivity for oil and organic solvents in mixed solvents. Furthermore, the low density and high porosity of silane-coated aerogels can make them an ideal absorbent for removing pollutants from water. A small piece of silane-coated composite NFAs was exposed to liquid–water mixtures, the dyed liquid was immediately absorbed by composite NFA (Fig. 9a, b, Movie S4 and 5), and the liquids-filler composite aerogel could hold the absorbed liquids without any liquids release. It is proposed that the silane-coated PVA-co-PE/SEP composite NFAs possess stiff pore walls, which could retain the absorbed liquids after absorption [52]. Furthermore, the pure oil could be collected from oil to water emulsions through a convenient pump method (Fig. S9 and Movie S6), demonstrating the potential applications of silane-coated PVA-co-PE/SEP composite NFAs in the field of oil–water separation.

Absorption of silane-coated PVA-co-PE/SEP composite NFAs (ρ = 12.57 mg cm−3): a diesel (dyed with Sudan red III) floating on water and b chloroform (dyed with Sudan red III) sinking below the water absorbed by silane-coated composite NFAs. c weight gain, d volume gain, e, f recyclability test of silane-coated PVA-co-PE/SEP composite NFAs: e cyclic evaporation test was applied to silane-coated PVA-co-PE/SEP composite NFAs for sorption of hexane, f cyclic squeezing test was used for silane-coated PVA-co-PE/SEP composite NFAs for sorption of phenoxin
Various types of liquids with different viscosity, density and polarity were used to investigate the oil/organic solvent absorbance of treated PVA-co-PE/SEP composite NFAs, and these liquids are common pollutants in both industrial and domestic areas. As shown in Fig. 9c, the silane-coated composite NFAs exhibited remarkable sorption performance for various types of liquids, the absorption capacities for liquids ranged from 45 to 108 times the weight of the dried silane-coated sample. This could be associated with low density, high porosity, superhydrophobicity and oleophilicity of the aerogels. The absorbency of silane-coated composite NFA was much better than that of previously reported absorbing materials [53,54,55].
The difference in viscosity between liquids has almost no effect on the absorption of the liquid [56]. However, it is obvious that the saturation absorption time of high viscosity liquids (diesel oil and soybean oil) is much longer than that of low viscosity liquids (chloroform and toluene) (Fig. S10). Low viscosity liquids could penetrate the 3D network structure of silane-coated composite NFAs more easily, in contrast to high viscosity liquids. In addition, it is clear that the absorbency of silane-coated composite NFAs depends strongly on the density of the liquid. For instance, the absorption capacity for phenoxin with a high density of 1.60 g cm−3 could reach 108 g/g, but the absorption capacity for hexane with a low density of 0.66 g cm−3 was just 45 g/g. Therefore, the absorption capacity might be related to the densities of various liquids, and the volume gain is shown in Fig. 9d. It was observed that values were approximately constant and large, ranged from 78.58 to 91.76%. And the values of silane-coated PVA-co-PE/SEP composite NFAs were obviously higher than that of pure PVA-co-PE NFAs (70.03–88.73%) [5].
The cyclic absorption capacity of the silane-coated PVA-co-PE/SEP composite NFAs is also investigated through evaporating and squeezing test, both of them could convert the liquid into available resources. The cyclic sorption–evaporation experiment was conducted by choosing hexane with a low boiling point (69 °C) as the adsorbate. After silane-coated composite, NFA was saturated with hexane, and the absorbed NFA was heated to a high temperature to release the steam of hexane. The steam of hexane was gathered by a condenser at the same time, and the composite NFA was recovered. As shown in Fig. 9e, the sorption–evaporation process was repeated for 10 times, and the silane-coated composite NFA showed a constant cyclic sorption capacity after 10 repetition tests. The cyclic sorption–squeezing test was demonstrated by selecting phenoxin as the absorbate. The absorbed NFA was squeezed to collect phenoxin. After the first cycle, the residual in NFA increased to approximate 19 times the original weight (Fig. 9f), which might be attributed to incomplete compression of silane-coated composite NFA [57]. The absorption capacity of silane-coated composite NFA also maintained stable after 10 repetition tests, because of their excellent mechanical properties. Therefore, both two methods could be adopted to recycle silane-coated composite NFA, and the PVA-co-PE/SEP composite NFA in this work denotes an ideal decontamination material for environmental protection applications.
Conclusion
We have demonstrated that composite nanofibrous aerogels (NFAs) could be fabricated through adding sepiolite nanorods (SEP) into the PVA-co-PE NFAs system. After introducing 30 wt% of SEP into aerogels, the density of composite NFAs decreased from 12.31 to 11.42 mg cm−3, and the porosity increased from 98.94 to 99.17%. With increasing SEP content, the surface hydrophilicity of composite NFAs increased. The compression strength of composite NFAs improved after introducing a small of SEP, while with additional addition of SEP, the compression strength decreased. The mechanical properties variation is shown to be controlled by a combination of stiffness of SEP and effective bonding of PVA-co-PE nanofibers. The hydrophobicity of composite NFAs improved after thermal chemical vapor deposition (TCVD) treatment. The surface of composite NFAs became more asperate, and siloxane nanoparticles became visible. The silane coating on the surface of composite NFAs could significantly improve the oil–water selectivity and separation efficiency of them. The deposition of siloxane nanoparticles also significantly improved compressive strength, durability, thermal stability and insulation of composite NFAs. The silane-coated PVA-co-PE/SEP composite NFAs exhibit a remarkable absorbency (mass-based absorption 45–108 g/g, volume-based absorption 78.58–91.76%) and sorption recyclability for various oils and organic solvents. We expected that the silane-coated composite NFAs prepared in this paper could provide prospective applications in alleviating some of the important energy and environmental issues.
References
Si Y, Yu JY, Tang XM, Ge JL, Ding B (2014) Ultralight nanofibre-assembled cellular aerogels with superelasticity and multifunctionality. Nat Commun 5:1–9
Zanini M, Lavoratti A, Lazzari LK, Galiotto D, Pagnocelli M, Baldasso C, Zattera AJ (2017) Producing aerogels from silanized cellulose nanofiber suspension. Cellulose 24:769–779
Ciftci D, Ubeyitogullari A, Huerta RR, Ciftci ON, Flores RA, Saldana MDA (2017) Lupin hull cellulose nanofiber aerogel preparation by supercritical CO2 and freeze drying. J Supercrit Fluid 127:137–145
Hayase G, Nonomura K, Hasegawa G, Kanamori K, Nakanishi K (2015) Ultralow-density, transparent, superamphiphobic boehmite nanofiber aerogels and their alumina derivatives. Chem Mater 27:3–5
Lu JW, Xu DD, Wei JK, Yan S, Xiao R (2017) Superoleophilic and flexible thermoplastic polymer nanofiber aerogels for removal of oils and organic solvents. ACS Appl Mater Interfaces 9:25533–25541
Fan BT, Chen SJ, Yao QF, Sun QF, Jin CD (2017) Fabrication of cellulose nanofiber/AlOOH aerogel for flame retardant and thermal insulation. Materials 10:1–10
Guo WW, Liu JJ, Zhang P, Song L, Wang X, Hu Y (2018) Multi-functional hydroxyapatite/polyvinyl alcohol composite aerogels with self-cleaning, superior fire resistance and low thermal conductivity. Compos Sci Technol 158:128–136
Zhou ZH, Zhang XX, Lu CH, Lan LD, Yuan GP (2014) Polyaniline-decorated cellulose aerogel nanocomposite with strong interfacial adhesion and enhanced photocatalytic activity. RSC Adv 4:8966–8972
Wu HJ, Chen YT, Chen QL, Ding YF, Zhou XQ, Gao HT (2013) Synthesis of flexible aerogel composites reinforced with electrospun nanofibers and microparticles for thermal insulation. J Nanomater 2013:1–8
Feng JZ, Zhang CR, Feng J, Jiang YG, Zhao N (2011) Carbon aerogel composites prepared by ambient drying and using oxidized polyacrylonitrile fibers as reinforcements. ACS Appl Mater Interfaces 3:4796–4803
Li LC, Yalcin B, Nguyen BN, Meador MAB, Cakmak M (2009) Flexible nanofiber-reinforced aerogel (xerogel) synthesis, manufacture, and characterization. ACS Appl Mater Interfaces 1:2491–2501
Wicklein B, Kocjan A, Salazar-Alvarez G, Carosio F, Camino G, Antonietti M, Bergstrom L (2015) Thermally insulating and fire-retardant lightweight anisotropic foams based on nanocellulose and graphene oxide. Nat Nanotechnol 10:277–283
Li YC, Schulz J, Mannen S, Delhom C, Condon B, Chang S, Zammarano M, Grunlan JC (2010) Flame retardant behavior of polyelectrolyte-clay thin film assemblies on cotton fabric. ACS Nano 4:3325–3337
Si Y, Fu Q, Wang X, Zhu J, Yu J, Sun G, Ding B (2015) Superelastic and superhydrophobic nanofiber-assembled cellular aerogels for effective separation of oil/water emulsions. ACS Nano 9:3791–3799
Ma HY, Tong LF, Xu ZB, Fang ZP (2008) Intumescent flame retardant-montmorillonite synergism in ABS nanocomposites. Appl Clay Sci 42:238–245
Chen HB, Wang YZ, Schiraldi DA (2014) Preparation and flammability of poly(vinyl alcohol) composite aerogels. ACS Appl Mater Interfaces 6:6790–6796
Losego MD, Grady ME, Sottos NR, Cahill DG, Braun PV (2012) Effects of chemical bonding on heat transport across interfaces. Nat Mater 11:502–506
Volle N, Challier L, Burr A, Giulieri F, Pagnotta S, Chaze AM (2011) Maya Blue as natural coloring fillers in a multi-scale polymer-clay nanocomposite. Compos Sci Technol 71:1685–1691
Dong YZ, Piao SH, Choi HJ (2018) Fe3O4/sepiolite magnetic composite particles and their magneto- responsive characteristics. Colloid Polym Sci 296:11–19
Tian G, Wang W, Li Z, Wang A (2017) MgO/palygorskite adsorbent derived from natural Mg-rich brine and palygorskite for high-efficient removal of Cd(II) and Zn(II) ions. J Environ Chem Eng 5:1027–1036
Ma Y, Zhang GK (2016) Sepiolite nanofiber-supported platinum nanoparticle catalysts toward the catalytic oxidation of formaldehyde at ambient temperature: efficient and stable performance and mechanism. Chem Eng J 288:70–78
Choy JH, Choi SJ, Oh JM, Park T (2007) Clay minerals and layered double hydroxides for novel biological applications. Appl Clay Sci 36:122–132
Wan CY, Chen BQO (2011) Synthesis and characterization of biomimetic hydroxyapatite/sepiolite nanocomposites. Nanoscale 3:693–700
Garcia-Lopez D, Fernandez JF, Merino JC, Santaren J, Pastor JM (2010) Effect of organic modification of sepiolite for PA 6 polymer/organoclay nanocomposites. Compos Sci Technol 70:1429–1436
Xie SB, Zhang SM, Wang FS, Yang MS, Seguela R, Lefebvre JM (2007) Preparation, structure and thermomechanical properties of nylon-6 nanocomposites with lamella-type and fiber-type sepiolite. Compos Sci Technol 67:2334–2341
Fukushima K, Tabuani D, Abbate C, Arena M, Ferreri L (2010) Effect of sepiolite on the biodegradation of poly(lactic acid) and polycaprolactone. Polym Degrad Stab 95:2049–2056
Huang DJ, Mu B, Wang AQ (2012) Preparation and properties of chitosan/poly (vinyl alcohol) nanocomposite films reinforced with rod-like sepiolite. Mater Lett 86:69–72
Can MF, Avdan L, Bedeloglu AC (2015) Properties of biodegradable PVA/sepiolite-based nanocomposite fiber mats. Polym Compos 36:2334–2342
Isci Y, Isci S (2017) Comparison of the clay minerals type on the properties of reinforced-PVA nanocomposites. Polym Compos 38:1698–1704
Zhang W, Zhang Y, Lu CH, Deng YL (2012) Aerogels from crosslinked cellulose nano/micro-fibrils and their fast shape recovery property in water. J Mater Chem 22:11642–11650
Zheng QF, Cai ZY, Gong SQ (2014) Green synthesis of polyvinyl alcohol (PVA)-cellulose nanofibril (CNF) hybrid aerogels and their use as superabsorbents. J Mater Chem A 2:3110–3118
Gerrard JA, Brown PK, Fayle SE (2002) Maillard crosslinking of food proteins I: the reaction of glutaraldehyde, formaldehyde and glyceraldehyde with ribonuclease. Food Chem 79:343–349
Ghosh A, Ali MA, Dias GJ (2009) Effect of cross-linking on microstructure and physical performance of casein protein. Biomacromolecules 10:1681–1688
Zhu J, Sun G (2014) Facile fabrication of hydrophilic nanofibrous membranes with an immobilized metal-chelate affinity complex for selective protein separation. ACS Appl Mater Interfaces 6:925–932
Deville S, Saiz E, Nalla RK, Tomsia AP (2006) Freezing as a path to build complex composites. Science 311:515–518
Deville S (2008) Freeze-casting of porous ceramics: a review of current achievements and issues. Adv Eng Mater 10:155–169
Visser CD, Perron G, Desnoyers JE (2011) The heat capacities and expansibilities of tert-butylalcohol-water mixtures from 6 to 65 °C. Can J Chem 55:856–862
Kasraian K, Deluca PP (1995) Thermal-analysis of the tertiary butyl alcohol-water system and its implications on freeze-drying. Pharmaceut Res 12:484–490
Gao YY, Gan HH, Zhang GK, Guo YD (2013) Visible light assisted Fenton-like degradation of rhodamine B and 4-nitrophenol solutions with a stable poly-hydroxyl-iron/sepiolite catalyst. Chem Eng J 217:221–230
Liu QZ, Chen JH, Mei T, He XW, Zhong WB, Liu K, Wang WW, Wang YD, Li MF, Wang D (2018) A facile route to the production of polymeric nanofibrous aerogels for environmentally sustainable applications. J Mater Chem A 6:3692–3704
Liang HW, Guan QF, Chen LF, Zhu Z, Zhang WJ, Yu SH (2012) Macroscopic-Scale template synthesis of robust carbonaceous nanofiber hydrogels and aerogels and their applications. Angew Chem Int Ed 51:5101–5105
Yang X, Cranston ED (2014) Chemically cross-linked cellulose nanocrystal aerogels with shape recovery and superabsorbent properties. Chem Mater 26:6016–6025
Xu M, Futaba DN, Yamada T, Yumura M, Hata K (2010) Carbon nanotubes with temperature-invariant viscoelasticity from-196 to 1000 °C. Science 330:1364–1368
Alvarez VA, Ruseckaite RA, Vazquez A (2003) Kinetic analysis of thermal degradation in poly(ethylenevinyl alcohol) copolymers. J Appl Polym Sci 90:3157–3163
Fernandez MD, Fernandez MJ (2008) Thermal decomposition of copolymers from ethylene with some vinyl derivatives. J Therm Anal Calorim 91:447–454
Koklukaya O, Carosio F, Waberg L (2017) Superior flame-resistant cellulose nanofibril aerogels modified with hybrid layer-by-layer coatings. ACS Appl Mater Interfaces 9:29082–29092
Pierre AC, Pajonk GM (2002) Chemistry of aerogels and their applications. Chem Rev 102:4243–4265
Jelle BP (2011) Traditional, state-of-the-art and future thermal building insulation materials and solutions—properties, requirements and possibilities. Energy Build 43:2549–2563
Wang X, Zhang H, Jana SC (2013) Sulfonated syndiotactic polystyrene aerogels: properties and applications. J Mater Chem A 1:13989–13999
Jimenez-Saelices C, Seantier B, Cathala B, Grohens Y (2017) Spray freeze-dried nanofibrillated cellulose aerogels with thermal superinsulating properties. Carbohydr Polym 157:105–113
Higginbotham AL, Lomeda JR, Morgan AB, Tour JM (2009) Graphite oxide flame-retardant polymer nanocomposites. ACS Appl Mater Interfaces 1:2256–2261
Jiang F, Hsieh YL (2014) Amphiphilic superabsorbent cellulose nanofibril aerogels. J Mater Chem A 2:6337–6342
Zhu Q, Chu Y, Wang ZK, Chen N, Lin L, Liu FT, Pan QM (2013) Robust superhydrophobic polyurethane sponge as a highly reusable oil-absorption material. J Mater Chem A 1:5386–5393
Zhang LH, Xu LD, Sun YL, Yang N (2016) Robust and durable superhydrophobic polyurethane sponge for oil/water separation. Ind Eng Chem Res 55:11260–11268
Yang LH, Bai B, Ding CX, Wang HL, Suo YR (2016) Synthesis and properties of the rapeseed meal-grafted-poly(methyl methacrylate-co-butyl acrylate) oil-absorbents. RSC Adv 6:9507–9517
Mulyadi A, Zhang Z, Deng YL (2016) Fluorine-free oil absorbents made from cellulose nanofibril aerogels. ACS Appl Mater Interfaces 8:2732–2740
Bi HC, Yin ZY, Cao XH, Xie X, Tan CL, Huang X, Chen B, Chen FT, Yang QL, Bu XY, Lu XH, Sun LT, Zhang H (2013) Carbon fiber aerogel made from raw cotton: a novel, efficient and recyclable sorbent for oils and organic solvents. Adv Mater 25:5916–5921
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 20874010) and the Program of Introducing Talents of Discipline to Universities (No. 111-2-04).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10853_2020_4400_MOESM5_ESM.avi
Movie S5: The chloroform (dyed with Sudan red III) was absorbed by silane-coated PVA-co-PE/SEP composite NFAs (AVI 653 kb)
Rights and permissions
About this article

Cite this article
Lu, J., Yan, S., Song, W. et al. Construction and characterization of versatile flexible composite nanofibrous aerogels based on thermoplastic polymeric nanofibers. J Mater Sci 55, 8155–8169 (2020). https://doi.org/10.1007/s10853-020-04400-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-04400-4