
Abstract
We report the properties of first synthesized ceramic samples of a perovskite solid solution (PbFe1/2Ta1/2O3)x[(PbMg1/3Nb2/3O3)0.7(PbTiO3)0.3]1−x, x = 0.4, 0.5. The solution is a single-phase material contrary to broadly studied multiphase (magnetic/ferroelectric) composites. Both compounds are ferroelectrics with a diffuse phase transition. An increase in x value results in a decrease in phase transition diffuseness, and an increase in the remnant polarization. 207Pb NMR shows that iron ions are nonuniformly distributed over octahedral sites and tend to random occupation of these sites in the perovskite structure. In particular, the NMR data indicate a tendency to form regions with a higher concentration of iron in the perovskite structure. Magnetic measurements show the coexistence of superparamagnetic and paramagnetic phases in the samples. The paramagnetoelectric (PME) coefficients determined by a dynamic method at room temperature have values β ≈ 0.15 × 10−15 s A−1 (x = 0.4) and 0.54 × 10−15 s A−1 (x = 0.5) at low magnetic fields (± 300 Oe), which are three thousands times larger than that in most single-phase magnetoelectric materials. Our measurements show that the main contribution to the PME response is caused by the superparamagnetic phase. Because the ME response is proportional to dM2/dH, it can be amplified by many orders of magnitude for the multiferroics with the superparamagnetic phase due to a sharp change of magnetization with the field.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A strong magnetoelectric (ME) coupling existing at room temperature is especially vital for novel functional device fabrication [1,2,3,4,5,6]. A straightforward way of increasing the magnitude of the ME response is to choose the components with large magnetostrictive and piezoelectric coefficients. For a long time, mainly composite multiferroics on the base of PbZr1−xTixO3 (PZT) or (PbMg1/3Nb2/3O3)0.7(PbTiO3)0.3 (PMN–PT) has been used. The latter component produces the largest ME effect and has been often used (see, e.g., [3]) in the multilayer multiphase (ferroelectric/ferromagnetic) structures. The reason is the large piezoelectricity of PMN–PT in the morphotropic region with the coexistence of the relaxor and ferroelectric phases. For a single crystal, it is 7 times larger than the piezoelectricity of PZT [6]. For ceramic materials, the piezoelectricity in PMN–PT is 2 times larger than that in PZT [7].
In the last years, considerable attention of scientists and engineers was paid to ferroelectric antiferromagnets PbFe1/2Ta1/2O3 (PFT), TN ≈ 130–180 K, and PbFe1/2Nb1/2O3 (PFN), TN ≈ 140 K [8, 9] and their solid solutions with PbZr0.53Ti0.47O3 [10,11,12,13,14,15,16,17]. Some of these solid solutions exhibit room-temperature multiferroism and large enough ME coupling, which includes a mixture of linear and biquadratic contributions. The ME coupling has been theoretically analyzed in Refs. [18, 19] and described by second and fourth rank tensors, namely, μijPiMj and ξijklPiPjMkMl (P is polarization and M is magnetization). The authors of Refs. [18, 19] have shown that large ME effect and the appearance of magnetization originate from the nanostructure of considered materials. Another mechanism of magnetization appearance in chemically disordered antiferromagnetic multiferroics was considered in Refs. [20, 21]. It was shown that antiferromagnetically interacting Fe3+ ions may form superstructures having a ferrimagnetic ground state. Such a structure has a different number of nonequivalent Fe positions in a unit cell. As a result, the ground state magnetization may reach several μB per Fe spin. Experimental indications on the ferrimagnetic superstructure formation were reported in Ref. [22] for PbFe1/2Sb1/2O3. The F-center exchange mechanism was proposed in Ref. [23]. It provides an explanation for the existence of ferromagnetism in oxides due to the presence of oxygen vacancies.
Recently the attention of scientists and engineers was attracted to the paramagnetoelectric (PME) effect introduced by Hou and Blombergen [24] described by term λijkPiMjMk. The results of experimental and theoretical studies of this effect in PFN and its solid solution with PbTiO3 were published in Refs. [25, 26].
It was not excluded that the replacement of PZT by PMN–PT in solid solution with PFT could lead to an essential increase in ME effect due to a larger piezoelectric coefficient in PMN–PT. Surprisingly, to the best of our knowledge, there is no information about the synthesis of the (PFT)x(PMN–PT)1−x solid solution. Perhaps the more complex characteristics of PMN–PT than PZT and thus the more complicated mechanism of ME effect were the main reason for the fact. On the other hand, both components of the (PFT)x(PMN–PT)1−x solid solution were broadly studied (see, e.g., [27,28,29,30,31,32,33]). In particular, it was shown that multiferroic PFT is antiferromagnetic–ferroelectric with ferroelectric phase transition at TC ≈ 250 K [31]. Considered PMN–PT is nonmagnetic with a maximum of dielectric permittivity at Tm ≈ 410–420 K [32, 33].
The aim of this study is to describe briefly the synthesis methodology of the novel single-phase multiferroic, to characterize the obtained samples and to present results of experimental and theoretical investigation of its properties, namely, polar, magnetic and magnetoelectric characteristics.
Samples preparation, structural characterization and experimental methods
Ceramic samples of (PFT)x(PMN0.7PT0.3)1−x were prepared by columbite precursor method [34] using the nanodispersed precursor powders of MgNb2O6 and TaFeO4 synthesized by the organonitrate method from chemicals with purity better than 99%. The received powders of MgNb2O6 and TaFeO4 were ground in an ethanol medium using a planetary mill with ZrO2 balls. After that, they were annealed for 6 h in corundum crucibles at 1050° and ground again for 6 h. Obtained nanodispersed TaFeO4 and MgNb2O6 were mixed in stoichiometric ratio with Ti(OH)4·nH2O and Pb(NO3)2, then ground in an ethanol medium for 6 h. A small amount of Li2CO3 (about 1 mol%) was added to the mixture. This addition promotes the formation of the perovskite modification of the solid solution and reduces its conductivity. The resulting powder was pressed into disks with a diameter of 13 mm and a height of 2.5 mm. The disks were calcined in corundum crucibles at temperatures from 950 to 1050 °C for 4 h.
Ceramic samples with x = 0.1, 0.2, 0.3, 0.4 and 0.5 were prepared for investigation. However, because the samples with x = 0.1, 0.2, 0.3 show only conventional paramagnetic behavior (see Fig. 10 in Appendix), only two compounds (x = 0.4 and 0.5) with multiferroic properties were chosen for detailed studies.
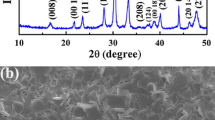
To confirm the perovskite structure of the synthesized samples, the X-ray diffraction patterns were obtained by using a DRON-UM1 diffractometer using monochromatic CuKα radiation (Fig. 1). XRD diffraction patterns of both samples correspond to the perovskites structure with minor addition of pyrochlore phase (less than 2.5 wt%). No magnetic impurities like Fe2O3, Fe3O4 or PbFe12O19 were detected by the XRD measurements.

XRD diffraction pattern of (PFT)0.4(PMN–PT)0.6 sample with pyrochlore reflexes marked by the asterisks
Polished and additionally thermally etched cross sections of the dense samples were analyzed by field emission scanning electron microscopy (FE-SEM) (Ultra plus, Carl Zeiss), and elemental analysis of the identified phases was performed with energy-dispersive spectroscopy (EDS), using Inca detector (Oxford Instrument). The samples were carbon coated prior to SEM/EDS analysis to ensure good conductivity during the investigation.
The EDS spectra of both compounds show the presence of all elements and confirm the chemical composition of these samples. The spectra were measured in ten different regions of each sample and have shown average homogeneity of the chemical composition within the error bars of the method (about 1 at.%), which indicates a formation of a single-phase solid solution of PMN–PT and PFT. Figure 2 shows the EDS spectrum of (PFT)0.4(PMN–PT)0.6 sample. The scanning electron microscopy image of this material is presented in the inset to Fig. 2. It shows the presence of micro-grains (2 to 5 μm) uniformly distributed over the surface of the sample. However, the sizes of coherent scatter regions calculated on the base of X-ray data (by the broadening of the diffraction lines) appeared to be about 70–85 nm for these samples, which indicates that the ceramic micro-grains consist of nanoscale structural units.

EDS spectrum of (PFT)0.4(PMN–PT)0.6 sample and the SEM micrograph of this sample
Dielectric properties of both compounds were measured on samples prepared in the form of tablets. These tablets were polished and coated with a silver conductive layer used as electrodes. Dielectric measurements were conducted using E7-20 impedance analyzer. The dielectric permittivity was measured at frequencies 25 Hz–200 kHz in the temperature range 290–480 K.
Nuclear magnetic resonance (NMR) measurements were performed using a commercial Bruker (Avance II) 400 MHz NMR spectrometer at 9.41 T. Extremely broad 207Pb NMR spectra were accumulated by a frequency-stepped method by acquiring spin echoes every 100 kHz. Individual echoes were Fourier transformed and superimposed in the frequency domain. A four-phase “exorcycle” phase sequence (xx, xy, x–x, x–y) was used to form echoes with minimal distortions due to antiechoes, ill-refocused signals and piezoresonances [35]. The length of the π/2 pulse in the spin echo sequence was tπ/2 = 4.4 μs, the spin echo delay time τ was 15 μs and the repetition time was 0.1 s.
The magnetization measurements were performed on the magnetometer with vibrating sample LDJ-9500 that is equipped by the regulating temperature system on the liquid nitrogen base.
The paramagnetoelectric coefficients were determined by a dynamic method [36] as a function of bias magnetic field Hdc at a small ac field hac = 0.64 Oe and frequency 311 Hz by measuring the current across the sample utilizing a high-sensitive lock-in-amplifier. High homogenous ac and dc magnetic fields were provided by a conventional EPR spectrometer. Both fields were applied normal to the surface of the sample. Before the measurements, the samples were poled at room temperature by applying a dc electric field of 10–15 kV/cm for 30 min. In every experiment, more than two runs were repeated with a reversed direction of Hdc, which results in a change of a sign of the signal. In this way, a possible spurious electromagnetic induction signal was distinguished from a true ME one whose sign is dependent on the PH product.
Experimental results
Investigation of dielectric properties
In this section, the dielectric measurements of multiferroic (PFT)x(PMN–PT)1−x (x = 0.4 and 0.5) and a discussion of the results will be presented.
The dependence of polarization on the electric field P(E) was measured at room temperature and shown in Fig. 3. The well-saturated hysteresis loops of both samples indicate the presence of a ferroelectric phase in these materials. One can see that an increase in the PFT content from x = 0.4 (04PFT) to x = 0.5 (05PFT) leads to an increase in the residual polarization from ~ 3 to ~ 4.4 µC cm−2 and to a decrease in the coercive field from ~ 3.5 to ~ 2.8 kV cm−1.

P–E hysteresis loops of (PFT)0.4(PMN–PT)0.6 (solid curve) and (PFT)0.5(PMN–PT)0.5 (dashed-dotted curve) solid solutions
Figure 4 shows the temperature variation of the dielectric permittivity measured for studied samples. Both samples have a wide maximum of the dielectric permittivity at temperature Tm, which value does not depend on the frequency, and is typical for ferroelectrics with a diffuse phase transition [37]. The temperature Tm decreases with increasing PFT content from 378 K for the 04PFT compound to 371 K for 05PFT. These temperatures approximately correspond to the transition temperatures expected for the solid solutions of PMN–PT and PFT: Tm = (1 − x)TPMN–PT + xTPFT, where TPMN–PT and TPFT are the transition temperatures of the components (see dashed line in the inset of Fig. 4). Some deviation from this expression can be explained by variations in a degree of ordering of Mg, Nb, Fe, Ti and Ta cations over “B” positions in the perovskite structure, which depends on the sample preparation and affects the phase transition temperature [38]. It is important to underline that the absence of any additional peaks in ε’(T) confirms the single-phase nature of the studied compounds.

Dependence of dielectric permittivity ε of (PFT)x(PMN–PT)1−x (x = 0.4 and 0.5) solid solutions on temperature at different frequencies. The inset shows the phase transition temperature as a function of the PFT content; experimental data for (PMN)0.7(PT)0.3 and PFT are taken from [39,40,41], respectively
As shown in Fig. 5, the temperature dependence of the inverse dielectric permittivity, measured for both samples at 1 kHz, does not follow the linear behavior predicted by the Curie–Weiss law for a certain temperature range above Tm. In this case, the modified Curie–Weiss law can be used to describe the temperature dependence of the dielectric permittivity [42]:

Temperature dependences of the reciprocal dielectric permittivity of the (PFT)x(PMN–PT)1−x (x = 0.4, 0.5) compounds; the inset shows logarithmic dependence of 1/ε − 1/εm on T − Tm
Parameter γ characterizes the degree of phase transition diffuseness, and its values lay in the region from 1 (for ferroelectrics) to 2 (for relaxors). Fitting of the experimental data gives the γ values equal to ~ 1.85 for 04PFT and ~ 1.8 for 05PFT. As shown in Ref. [37], the diffuse phase transition could be considered as an intermediate state in which both ferroelectric and relaxor phases can be presented simultaneously. This allows us to suggest that both relaxor and normal ferroelectric phases coexist in the studied compounds, and an increase in the PFT content leads to an increase in the ferroelectric phase contribution, which is in agreement with the higher remnant polarization measured for 05PFT as shown in Fig. 3.
207Pb nuclear magnetic resonance
207Pb Nuclear Magnetic Resonance (NMR) spectra of PFT and the solid solutions (PFT)x(PMN–PT)1−x (x = 0.4, 0.5) are presented in Fig. 6. The spectra reflect magnetic interactions of Fe3+ ions responsible for magnetic properties of studied materials with the nuclei of Pb ions giving the main contribution to the polarization of these materials. Usually, 207Pb NMR spectra of perovskites comprising iron in their structure [43, 44] consist of a single featureless line (800 kHz–1 MHz wide) due to a distribution of superhyperfine interactions between 207Pb and Fe3+ presented in these samples. It is important that an analysis of spectra of studied solid solutions provides an opportunity to determine the distribution of Fe atoms over “B” sites in the perovskite structure. For better resolution of spectrum, the sample 04PFT was heated up to 450 K, to observe the narrowing of lines due to a motional averaging of the interactions, arising as a result of fast thermal reorientations of magnetic moments. This temperature is also much higher than the temperature of ferroelectric to paraelectric phase transition (~ 380 K, see Fig. 4), which reduces the effects associated with distribution of superhyperfine interactions due to a decrease in the spread of Fe–Pb distances, and well higher than the estimated temperature range of the superparamagnetic phase existence (Tc ≈ 400 K, see “Magnetization” section), which ensures that the entire sample is in a paramagnetic state and all Pb atoms are visible in the NMR spectrum.

207Pb NMR spectra of PFT, solid solutions (PFT)x(PMN–PT)1−x (x = 0.4, 0.5) at room temperature and (PFT)0.4(PMN–PT)0.6 at 450 K
The largest influence on the frequency shift of 207Pb NMR spectra has the Fermi contact interaction, which transferred a Fe3+ magnetization to Pb nucleus due to the chemical bond between these atoms. This interaction decreases quickly with the number of bonds involved and the main contribution to the spectra originates from nearest-neighbor Fe3+ ions. Thus, the spin-Hamiltonian for the sample being in the paramagnetic state can be presented in the form:
where μ0H0 ~ 9.4 T is the external magnetic field, γN is the gyromagnetic ratio of 207Pb nuclei, 〈S〉 is the thermally averaged spin of Fe3+, A is the Fermi contact interaction constant, and N is the number of nearest-neighbor Fe ions in Pb surroundings.
As shown in Fig. 6, 207Pb NMR spectra of 04PFT and 05PFT compounds can be represented as a convolution of two lines (spectral components), which indicates a nonuniform distribution of Fe atoms. This is quite obvious for (PFT)0.5(PMN–PT)0.5 with 25% of all “B” sites occupied by Fe. In the case of uniform distribution, each of the Pb atoms (surrounded by eight “B” sites) will have exactly two nearest-neighbor iron atoms (and four in the case of PFT), which should lead to a single line in the NMR spectrum of this compound.
The relative intensities of the abovementioned two lines indicate a close to a random distribution of Fe atoms over the “B” sites in the perovskite structure. Indeed, in the case of a random distribution of iron atoms, the probability of the simultaneous location of N iron atoms in the nearest environment of a lead can be expressed as:
where p is a probability of finding an iron atom in the nearest environment of lead. For 04PFT (p = 0.2) this gives P(0) = 0.168, P(1) = 0.336, P(2) = 0.294, P(3) = 0.147 and P(4) = 0.046. Taking into account that due to the superhyperfine interaction, the shift of a particular spectrum ∆ν = –NA〈S〉/h depends on the number of nearest-neighbor Fe ions, the spectrum from all 207Pb nuclei should consist of five lines located at distances A〈S〉/h from each other, whose relative integral intensities equal to P(N) and whose width depends on the number of neighboring iron atoms. Analysis of the spectrum for this compound shows that the relative integral intensities of two lines obtained after fitting are close to P(0) + P(1) = 0.504 and P(2) + P(3) + P(4) = 0.487 (see Table 1). This allows us to suppose that the narrow line corresponds to lead atoms having from 0 to 1 nearest neighboring iron atoms, and the wide line to lead with a larger number of nearest neighboring iron atoms.
The relative integral intensities obtained for 05PFT also fit the assumption of a close to a random distribution of iron atoms (see Table 1), but higher temperature range of the superparamagnetic phase existence (~ 490 K) does not allow to reliably overreach this temperature in order to obtain the paramagnetic state of the entire sample.
In a case of arbitrary distribution of Fe atoms in PFT, the probability of B sites occupation by Fe is 0.5. Thus, the probability of the location of 0 or 1 Fe atoms in the nearest environment of Pb is negligibly small [see Eq. (1)], and a much more essential contribution arises from Pb having from two to six nearest neighboring iron atoms. Overlapping of these lines (extremely broadened by the magnetic interactions between Pb and Fe atoms) would results in only one wide line in the 207Pb NMR spectrum of PFT. Thus, the 207Pb NMR spectra of PFT cannot be used for the determination of iron atoms distribution.
Thereby, 207Pb NMR shows that iron atoms tend to random occupation of the sites “B” in the perovskite structure. At the same time, the intensity of the line corresponding to Pb with a higher number of nearest-neighbor Fe ions is noticeably larger than it follows from Eq. (1) (see Table 1). This allows us to suppose the existence of some tendency to form areas with a higher concentration of iron.
Magnetic properties
The magnetic response of solid solutions (PFT)x(PMN–PT)1−x is due to the presence of octahedrally coordinated Fe3+ ions having 3d5 electronic d-shell configuration in S-state, spin SFe =5/2 and g-factor g ≈ 2. Their fraction in the formula unit is x/2. Figure 7 shows magnetization isotherms M(H) at T = 120, 193, 293 K for two samples. The M(H) isotherms have a qualitatively similar look for x = 0.4 and 0.5. For all considered temperatures, the curves are anhysteretic, i.e., reversible. We see that the measured magnetization may be presented as a sum of a paramagnetic contribution that is proportional to the field Mp(H, T) = χp(T)H and of superparamagnetic-like contribution Ms(H, T) that saturates at the field of the order of 1 kOe. The absence of noticeable hysteresis for observed curves allows considered nonparamagnetic contributions as superparamagnetic ones registered at T ≫ Tb, Tb being a blocking temperature [45, 46]. Therefore, the blocking temperature is Tb <120 K.

The solid lines in Fig. 7 show the fit of M(H) curves by the sum of paramagnetic and superparamagnetic contributions
where Ns is the number of magnetic moments μs(T) in 1 g of the material; L(x) is the Langevin function \( L(x) \equiv \coth(x) - 1/x \). The parameters of the fit are given in Table 2. The details of the fitting procedure are given in the Appendix [Eqs. (6, 7)].
The susceptibility χp(T) is determined by the slope of the curve at large H and approximated by Curie–Weiss law (see Fig. 8)

Inverse magnetic susceptibility of the (PFT)x(PMN–PT)1−x solid solutions with x = 0.4 (squares) and x = 0.5 (triangles). The lines show the Curie–Weiss law fit by Eq. (2)
where the paramagnetic Curie–Weiss temperature TCW is the measure of average interaction between the spins [47,48,49]; Cp is the Curie constant. The negative sign of TCW ≈ − 80 K means that the average interaction between spins in paramagnetic regions of our samples has AFM sign.
Magnetoelectric measurements
In our experiment, the ME effect is manifested as a polarization Pac induced by a small ac magnetic field hac under application of dc field Hdc [24,25,26]. We use the standard notations for ME coefficients that are given by the expansion [see Eqs. (8)–(20) of the Appendix]
where P0 is the spontaneous polarization; αij is a linear coupling, which is absent in the paramagnetic phase, βijk is the PME coupling coefficient.
With using collinear dc and ac magnetic fields H = Hdc + hac sin ωt, the first harmonic of the ac polarization detected by lock-in detector is
Here β(T) is the paramagnetoelectric susceptibility. In ceramics, β(T) represents an average of the different elements of the tensor
where F(E,H) [6] is the free energy density.
The PME response is studied by measuring the current across the sample. More precisely, the PME current is determined from Eq. (4) as
with the lock-in (phase) detection at the frequency ω. Here σ is the area of the sample.
Figure 9 shows ME current as a function of the applied dc magnetic field in a PFT–PMN–PT ceramics for two compositions x = 0.4 and 0.5.

ME current amplitude in (PFT)x(PMN–PT)1−x as a function of applied dc magnetic field for ax = 0.4 measured at 293 K and bx = 0.5 measured at 293 K and 120 K. Room temperature value of the paramagnetoelectric coefficient β is indicated. The dash line in (b) shows a “linear” part of the ME current from paramagnetic component. Inset: ME current amplitude for x = 0.4 superimposed with dM2/dH calculated from the data of Fig. 7a
One can see that in both samples, ME signal sharply increases with an increase in the dc magnetic field in the range of ± 300 Oe, then it saturates in value and decreases down to almost zero at fields larger ± 3000 Oe. Inset in Fig. 9a shows that ME response correlates well with the dM2/dH for the M(H) curves presented in Fig. 7 (see the derivation of Eq. (20) in Appendix for arguments in favor of this correlation). The ME current does not change too much with temperature lowering down to 120 K (Fig. 9b). Only the current peak position moves from ± 300 to ± 400 Oe at 120 K.
In the field range |H| < 300 Oe, the slope of the curves allows us to extract the coefficient β via the formula (5). This results in β ≈ 0.15 × 10−15 s A−1 for x = 0.4 and β ≈ 0.54 × 10−15 s A−1 for x = 0.5 in (PFT)x(PMN–PT)1−x, which are about three orders of magnitude larger than those measured in conventional magnetoelectric materials at room temperatures (see “Discussion” section).
Our measurements show that the main contribution to the ME current is caused by the superparamagnetic phase while the contribution of isolated Fe3+ spins from the paramagnetic phase is negligibly small due to their much lower magnetic moment. The “linear” part of the ME response from the paramagnetic phase is illustrated by the dashed line in Fig. 9b. For the sample with lower Fe concentration (x = 0.4), this contribution is practically invisible.
Discussion
Let us discuss in more detail the ME effect in the PFT–PMN–PT solid solution. It would be interesting to compare the measured PME coefficients with those obtained by the same method for other multiferroics. The authors of Refs. [25, 26] have investigated solid solution (PbFe1/2Nb1/2O3)x(PbTiO3)1−x (PFN–PT). The structure, properties and phase diagram of PFN are similar to those of PFT; however, the properties of the second components of solid solutions, namely (PMN)0.7(PT)0.3 and PT [25], are completely different. One can suppose that the difference in the ME effect is mainly induced by the second component of the solid solution. The ME coefficient measured by us at room temperature is equal to β ≈ 0.54 × 10−15 s A−1 (x = 0.5) and is related to superparamagnetic component in the PFT–PMN–PT solid solution, while that coefficient obtained for PFN-PT [25, 26] appeared to be much smaller, β ~ 10−18 s A−1 and is related to paramagnetic component which dominates over a superparamagnetic impurity. The superpaparamagnetic contribution is not even visible in magnetic measurements for PFN–PT (see Fig. 1d of Ref. [25]). The obtained values of the PME coefficient appeared to be also much larger than that determined in several other antiferromagnetic multiferroic materials (see Table I in Ref. [26]): BiFeO3 (2.1 × 10−19 s A−1) Gd2(MoO4)3 (0.8 × 10−18 s A−1) and NiSO4·6H2O (0.7 × 10−18 s A−1).
Note that the strong scattering of ME effect values in PFT–PZT solid solutions (see, e.g., [10, 12,13,14] made it cumbersome to perform a direct comparison with our results obtained for PFT–PMN–PT solid solution.
In some works [50,51,52], the slim hysteresis loops in perovskite ceramic samples are ascribed to a small amount of ferro- or ferri- magnetic impurity phases. The presence of such impurities in our samples would imply that they are, in fact, 0–3 type two-phase composites (see, e.g., review paper [53]). However, a small undetectable by XRD fraction of the magnetic impurity phase (see estimates in Appendix) can not produce appreciable ME response for two-phase composite material as the ME signal sharply goes to zero at approaching zero fraction volume of the magnetic component [53].
Our study demonstrates that multiferroics with superparamagnetic and ferroelectric phases can be considered as promising materials for applications along with composite multiphase (ferroelectric/ferromagnetic) structures. Because the ME response in multiferroics with the superparamagnetic phase is proportional to dM2/dH, its value can be amplified by many orders due to a sharp change of magnetization with the field.
Conclusions
The solid solutions (PbFe1/2Ta1/2O3)x[(PbMg1/3Nb2/3O3)0.7(PbTiO3)0.3](1−x) = (PFT)x(PMN–PT)(1−x), x = 0.4, 0.5 have been synthesized. XRD diffraction patterns of investigated samples correspond to the perovskite structure with minor addition of pyrochlore phase (less than 2.5 wt%). The EDS analysis reveals the homogeneity of the chemical composition and indicates the formation of a single-phase solid solution. Dielectric measurements show that both materials are ferroelectrics with a diffuse phase transition with the spontaneous polarization of about 5 µC cm−2. An increase in the PFT content leads to an increase in the relative volume of the ferroelectric phase. Temperature dependence of the dielectric permittivity also confirms that both studied compounds are single-phase solid solutions (PFT)x(PMN–PT)1−x. 207Pb NMR shows that iron atoms tend to random occupation of the sites “B” in the perovskite structure. Analysis of 207Pb NMR data reveals a tendency to form areas with a higher concentration of iron.
Magnetic properties measurements manifest a superparamagnetic behavior of the samples.
The magnetoelectric coefficients are determined by the dynamic method. Their low-field values at room temperature β ≈ 0.15 × 10−15 s A−1 and 0.54 × 10−15 s A−1, respectively, for samples with x = 0.4 and x = 0.5 are three orders of magnitude larger than that in conventional antiferromagnetic multiferroics. Our measurements and theoretical analysis show that the main contribution to the ME current is caused by the superparamagnetic phase.
References
Spaldin NA, Ramesh R (2008) Electric-field control of magnetism in complex oxide thin films. MRS Bull 33:1047–1050
Scott JF (2012) Applications of magnetoelectrics. J Mater Chem 22:4567–4574
Pyatakov AP, Zvezdin AK (2012) Magnetoelectric and multiferroic media. Physics Uspekhi 55:557–581
Cherifi RO, Ivanovskaya V, Phillips LC, Zobelli A, Infante IC, Jacquet E, Garcia V, Fusil S, Briddon PR, Guiblin N, Mougin A, Ünal AA, Kronast F, Valencia S, Dkhil B, Barthélémy A, Bibes M (2014) Electric-field control of magnetic order above room temperature. Nat Mater 13:345–351
Scott JF (2013) Room-temperature multiferroic magnetoelectrics. NPG Asia Mater 5(11):e72
Feibig M (2005) Revival of the magnetoelectric effect. J Phys D Appl Phys 38:R123–R152
Ryu J, Priya S, Uchino K, Kim H-E (2002) Magnetoelectric effect in composites of magnetostrictive and piezoelectric materials. J Electroceram 8:107–119
Smolenskii G, Loffe VA (1958) Communication No 71. Colloque International du Magnetisme, Grenoble
Bokov V, Myl’nikova I, Smolenskii GA (1962) Ferroelectric antiferromagnetics. Sov Phys JETP 15:447–449
Sanchez DA, Ortega N, Kumar A, Roque-Malherbe R, Polanco R, Scott JF, Katiyar RS (2011) Symmetries and multiferroic properties of novel room-temperature magnetoelectrics: lead iron tantalate–lead zirconate titanate (PFT/PZT). AIP Adv 1:042169
Martin LW, Ramesh R (2012) Multiferroic and magnetoelectric heterostructures. Acta Mater 60:2449–2470
Evans DM, Schilling A, Kumar A, Sanchez D, Ortega N, Arredondo M, Katiyar RS, Gregg JM, Scott JF (2013) Magnetic switching of ferroelectric domains at room temperature in multiferroic PZTFT. Nat Commun 4:1534
Sanchez DA, Ortega N, Kumar A, Sreenivasulu G, Katiyar RS, Scott JF, Evans DM, Arredondo-Arechavala M, Schilling A, Gregg JM (2013) Room-temperature single phase multiferroic magnetoelectrics: Pb(Fe, M)x(Zr, Ti)(1–x)O3 [M = Ta, Nb]. J Appl Phys 113:074105
Evans DM, Schilling A, Kumar A, Sanchez D, Ortega N, Katiyar RS, Scott JF, Gregg JM (2014) Switching ferroelectric domain configurations using both electric and magnetic fields in Pb(Zr, Ti)O3–Pb(Fe, Ta)O3 single-crystal lamellae. Philos Trans R Soc A 372:20120450
Schiemer J, Carpenter MA, Evans DM, Gregg JM, Schilling A, Arredondo M, Alexe M, Sanchez D, Ortega N, Katiyar RS, Echizen M, Colliver E, Dutton S, Scott JF (2014) Studies of the room-temperature multiferroic Pb(Fe0.5Ta0.5)0.4(Zr0.53Ti0.47)0.6O3: resonant ultrasound spectroscopy, dielectric, and magnetic phenomena. Adv Funct Mater 24:2993–3002
Schiemer JA, Lascu I, Harrison RJ et al (2016) Elastic and anelastic relaxation behaviour of perovskite multiferroics I: PbZr0.53Ti0.47O3 (PZT)–PbFe0.5Nb0.5O3 (PFN). J Mater Sci 51:10727–10760. https://doi.org/10.1007/s10853-016-0280-2
Schiemer JA, Lascu I, Harrison RJ et al (2017) Elastic and anelastic relaxation behaviour of perovskite multiferroics II: PbZr0.53Ti0.47O3 (PZT)–PbFe0.5Ta0.5O3 (PFT). J Mater Sci 52:285–304. https://doi.org/10.1007/s10853-016-0330-9
Glinchuk MD, Eliseev EA, Morozovska AN (2014) Novel room temperature multiferroics on the base of single-phase nanostructured perovskites. J Appl Phys 116:054101
Glinchuk MD, Eliseev EA, Morozovska AN (2016) Landau–Ginzburg description of anomalous properties of novel room temperature multiferroics Pb(Fe1/2Ta1/2)x(Zr0.53Ti0.47)1−xO3 and Pb(Fe1/2Nb1/2)x(Zr0.53Ti0.47)1−xO3. J Appl Phys 119:024102
Kuzian RO, Laguta VV, Richter J (2014) Lieb–Mattis ferrimagnetic superstructure and superparamagnetism in Fe-based double perovskite multiferroics. Phys Rev B 90:134415
Kuzian RO, Richter J, Kuz’min MD, Hayn R (2016) Lieb–Mattis ferrimagnetism in magnetic semiconductors. Phys Rev B 93:214433
Laguta VV, Stephanovich VA, Savinov M et al (2014) Superspin glass phase and hierarchy of interactions in multiferroic PbFe1/2Sb1/2O3: an analog of ferroelectric relaxors? New J Phys 16:113041
Coey JMD, Douvalis AP, Fitzgerald CB, Venkatesan M (2004) Ferromagnetism in Fe-doped SnO2 thin films. Appl Phys Lett 84:1332–1334
Hou SL, Bloembergen N (1965) Paramagnetoelectric effects in NiSO4·6H2O. Phys Rev 138:A1218–A1226
Laguta VV, Morozovska AN, Eliseev EA, Raevski IP, Raevskaya SI, Sitalo EI, Prosandeev SA, Bellaiche L (2016) Room-temperature paramagnetoelectric effectin magnetoelectric multiferroics Pb(Fe1/2Nb1/2)O3 and its solid solution with PbTiO3. J Mater Sci 51:5330–5342. https://doi.org/10.1007/s10853-016-9836-4
Laguta VV, Stephanovich VA, Raevski IP, Raevskaya SI, Titov VV, Smotrakov VG, Eremkin VV (2017) Magnetoelectric effect in antiferromagnetic multiferroic Pb(Fe1/2Nb1/2)O3 and its solid solutions with PbTiO3. Phys Rev B 95:014207
Falqui A, Lampis N, Geddo-Lehmann A, Pinna G (2005) Low-temperature magnetic behavior of perovskite compounds PbFe1/2Ta1/2O3 and PbFe1/2Nb1/2O3. J Phys Chem 109:22967–22970
Choudhury RNP, Rodriguez C, Bhattacharya P, Katiyar RS, Rinaldi C (2007) Low-frequency dielectric dispersion and magnetic properties of La, Gd modified Pb(Fe1/2Ta1/2)O3 multiferroics. J Magn Magn Mater 313:253–260
Bharti C, Dutta A, Shannigrahi S, Sinha TP (2012) Electronic structure, magnetic and electrical properties of multiferroic PbFe1/2Ta1/2O3. J Magn Magn Mater 324:955–960
Lampis N, Franchini C, Satta G, Geddo-Lehmann A, Massidda S (2004) Electronic structure of PbFe1/2Ta1/2O3: crystallographic ordering and magnetic properties. Phys Rev B 69:064412
Zhu WZ, Kholkin A, Mantas PQ, Baptista JL (2000) Preparation and characterisation of Pb(Fe1/2Ta1/2)O3 relaxor ferroelectric. J Eur Ceram Soc 20:2029–2034
Choi SW, Shrout RTR, Jang SJ, Bhalla AS (1989) Dielectric and pyroelectric properties in the Pb(Mg1/3Nb2/3)O3–PbTiO3 system. Ferroelectrics 100(1):29–38
Feng Z, Zhao X, Luo H (2006) Composition and orientation dependence of dielectric and piezoelectric properties in poled Pb(Mg1/3Nb2/3)O3–PbTiO3 crystals. J Appl Phys 100:024104
Swartz SL, Shrout TR (1982) Fabrication of perovskite lead magnesium niobate. Mater Res Bull 17:1245–1250
Ernst R, Bodenhausen S, Wokaun A (1987) Principles of NMR in One and two dimensions. Oxford University Press, New York
Kumar MM, Srinivas A, Surynarayana SV, Kumar GS, Bhimasankaram T (1998) An experimental setup for dynamic measurement of magnetoelectric effect. Bull Mater Sci 21:251–255
Shvartsman VV, Kleemann W, Dec J, Xu ZK, Lu SG (2006) Diffuse phase transition in BaTi1−xSnxO3 ceramics: an intermediate state between ferroelectric and relaxor behavior. J Appl Phys 99:124111
Bogs M, Beige H, Pitzius P, Schmitt H (1992) Linear and nonlinear dielectric, elastic and electromechanical properties of Pb(Sc1/2Ta1/2)O3 ceramics. Ferroelectrics 126(1):197–202
Choi SW, Shrout TR, Jang SJ, Bhalla AS (1989) Morphotropic phase boundary in Pb(Mg1/3Nb2/3)O3–PbTiO3 system. Mater Lett 8(6–7):253–255
Geddo-Lehmann A, Sciau P (1999) Ferroelastic symmetry changes in the perovskite PbFe0.5Ta0.5O3. J Phys Condens Matter 11:1235–1245
Nomura S, Takabayashi H, Nakagawa T (1968) Dielectric and magnetic properties of Pb(Fe1/2Ta1/2)O3. Jpn J Appl Phys 7:600–604
Santos IA, Eiras JA (2001) Phenomenological description of the diffuse phase transition in ferroelectrics. J Phys Condens Matter 13:11733–11740
Zagorodniy YuO, Kuzian RO, Kondakova IV, Maryško M, Chlan V, Štěpánková H, Olekhnovich NM, Pushkarev AV, Radyush YuV, Raevski IP, Zalar B, Laguta VV, Stephanovich VA (2018) Chemical disorder and 207Pb hyperfine fields in the magnetoelectric multiferroic Pb(Fe1/2Sb1/2)O3 and its solid solution with Pb(Fe1/2Nb1/2)O3. Phys Rev Mater 2:014401
Kouřil K, Chlan V, Štĕpánková H, Řezníček R, Laguta VV, Raevski IP (2015) NMR study of multiferroic iron niobate perovskites. Acta Phys Pol A 127(2):234–236
Bedanta S, Kleemann W (2009) Supermagnetism. J Phys D Appl Phys 42:013001
Wiekhorst F, Shevchenko E, Weller H, Kötzler J (2003) Anisotropic superparamagnetism of monodispersive cobalt–platinum nanocrystals. Phys Rev B 67:224416
Opechowski W (1937) On the exchange interaction in magnetic crystals. Physica 4:181
Schmidt H-J, Lohmann A, Richter J (2011) Eighth-order high-temperature expansion for general Heisenberg Hamiltonians. Phys Rev B 84:104443
Lohmann A, Schmidt H-J, Richter J (2014) Tenth-order high-temperature expansion for the susceptibility and the specific heat of spin-Heisenberg models with arbitrary exchange patterns: application to pyrochlore and kagome magnets. Phys Rev B 89:014415
Raevski IP, Titov VV, Malitskaya MA et al (2014) Studies of ferroelectric and magnetic phase transitions in multiferroic PbFe0.5Ta0.5O3–PbTiO3 solid solution ceramics. J Mater Sci 49:6459–6466. https://doi.org/10.1007/s10853-014-8376-z
Raevski IP, Molokeev MS, Misyul SV et al (2015) Studies of ferroelectric and magnetic phase transitions in multiferroic PbFe0.5Ta0.5O3. Ferroelectrics 475(1):52–60
Cho SY, Kim JS, Jang MS (2006) Dielectric, ferroelectric and ferromagnetic properties of 0.8PbFe0.5Ta0.5O3–0.2PbTiO3 ceramics. J Electroceram 16:369–372
Nan CW, Bichurin MI, Dong S, Viehland D, Srinivasan G (2008) Multiferroic magnetoelectric composites: historical perspective, status, and future directions. J Appl Phys 103:031101
Laguta V, Rosa J, Jastrabik L, Blinc R, Cevc P, Zalar B, Remskar M, Raevskaya S, Raevski I (2010) 93Nb NMR and Fe3+ EPR study of local magnetic properties of magnetoelectric Pb(Fe1/2Nb1/2)O3. Mater Res Bull 45:1720–1727
Battle PD, Gibb T, Herod A, Kim S-H, Munns P (1995) Investigation of magnetic frustration in A2FeMO6 (A = Ca, Sr, Ba; M = Nb, Ta, Sb) by magnetometry and Mössbauer spectroscopy. J Mater Chem 5:865–870
Battle PD, Gibb T, Herod A, Hodges J (1995) Sol–gel synthesis of the magnetically frustrated oxides Sr2FeSbO6 and SrLaFeSnO6. J Mater Chem 5:75–78
Blinc R, Laguta VV, Zalar B, Zupancic B, Itoh M (2008) O17 and 93Nb NMR investigation of magnetoelectric effect in Pb(Fe1/2Nb1/2)O3. J Appl Phys 104:084105
Kuzian RO, Kondakova IV, Dare AM, Laguta VV (2014) Magnetic interactions in disordered perovskite PbFe1/2Nb1/2O3 and related compounds: dominance of nearest-neighbor interaction. Phys Rev B 89:024402
Raevski IP, Kubrin SP, Raevskaya SI, Sarychev DA, Prosandeev SA, Malitskaya MA (2012) Magnetic properties of PbFe1/2Nb1/2O3: Mössbauer spectroscopy and first-principles calculations. Phys Rev B 85:224412
Diaz S, Suárez N, Faloh J, Sánchez J, Leccabue F et al (1997) Magnetic properties of PbFe12O19 hexaferrites powders obtained by metallorganic decomposition. Le Journal de Physique IV 07(C1):C1-333–C1-334
Yang N, Yang H, Jia J, Pang X (2007) Formation and magnetic properties of nanosized PbFe12O19 particles synthesized by citrate precursor technique. J Alloys Compd 438:263–267
Chaudhury S, Rakshit SK, Parida SC et al (2008) Studies on structural and thermo-chemical behavior of MFe12O19(s) (M = Sr, Ba and Pb) prepared by citrate–nitrate gel combustion method. J Alloys Compd 455:25–30
Bezlepkin AA, Kuntsevich SP, Kostyukov VI (2015) Orientational and relaxation features of the dynamic magnetic susceptibility of PbFe12O19 upon transition from the magnetically ordered to paramagnetic state. Phys Solid State 57:2213–2216
Pullar RC (2012) Hexagonal ferrites: a review of the synthesis, properties and applications of hexaferrite ceramics. Prog Mater Sci 57:1191–1334
Albanese G, Díaz-Castañón S, Leccabue F, Watts BE (2000) Mössbauer and magnetic investigation of scandium and indium substituted PbFe12O19 hexagonal ferrite. J Mater Sci 35:4415–4420. https://doi.org/10.1023/A:1004869310024
Maryško M, Frait Z, Krupička S (1997) FMR and static magnetic properties of gallium substituted magnetoplumbite. J. Phys. IV (France) 7:C1-347–C1-348
Perekalina TM, Vinnik MA, Zvereva RI, Shchurova AD (1971) Magnetic properties of hexagonal ferrites with weak exchange coupling between sublattices. Sov Phys JETP 32:813–814
Glinchuk MD, Eliseev EA, Morozovska AN, Blinc R (2008) Giant magnetic effect induced by intrinsic surface stress in ferroic nanorods. Phys Rev B 77:024106
Acknowledgements
We thank A. N. Morozovska and E. A. Eliseev for critical remarks and A. E. Likhtin for technical assistance.
Funding
Funding was provided by National Academy of Sciences of Ukraine (Projects III-8-16 and 44/18-H).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix
Appendix
Magnetization analysis
The solid solutions are materials with perovskite structure ABO3. We may rewrite the chemical formula as (PFT)x(PMN–PT)1−x = PbFex/2Tax/2Mg0.7(1−x)/3Nb1.4(1−x)/3Ti0.3(1−x)O3. Magnetic ions Fe and nonmagnetic M = Ta, Mg, Nb, Ti occupy the sites of cubic sublattice B with the percentage given by the corresponding subscript in the formula. The Fe ions may be distributed randomly or form some kind of short- or long-range chemical order. Evidence for partial chemical ordering in the B sublattice of complex perovskites comes from experiments [54,55,56,57] and theory [20,21,22, 58, 59]. Our NMR studies (see above) confirm the nonuniform distribution of Fe in the B sublattice.
Magnetic measurements for (PFT)x(PMN–PT)1−x with x = 0.1, 0.2, 0.3 (Fig. 10) show that these samples are in the paramagnetic state in the studied temperature range. Thus, we omitted any additional investigations of these samples and focused our attention on samples with x = 0.4 and 0.5.

Magnetization dependence on magnetic field for solid solution (PFT)x(PMN–PT)1−x for x = 0.1 (a), 0.2 (b) and 0.3 (c) at different temperatures
We assume that a part of Fe3+ ions xFe < x/2 contribute to the paramagnetic response χp(T), another part, xs= x/2 − xFe, participates in superparamagnetic response. The Curie constant
gives the number Np of Fe spins (per 1 g of the material) in the paramagnetic state; μB is Bohr magneton. Figure 8 shows that these numbers are very close for both samples Np ≈ 2.8 × 1020. Using the molar mass of the solid solution mM(x), we find xFe = mM(x)Np/NA ≈ 0.16 for both samples, NA is the Avogadro number.
After that, we fit M(H) curves by the sum of paramagnetic and superparamagnetic contributions (cf. Eq.~(3) from Ref. [45]) given by Eq. (2).
We assume that the distribution of superspin moments is sharply peaked at the value μs(T) (i.e., all superspins are equal). Noting that L(x) → 1, for x → ∞, and \( \left. {\frac{\partial L(x)}{\partial x}} \right|_{x = 0} = \frac{1}{3} \), we may obtain the superspin moment value, and the number of superspins
from the slope of the superparamagnetic contribution to the isotherm curve at the origin \( \left. {\frac{{\partial M_{s} \left( {H,T} \right)}}{\partial H}} \right|_{H = 0} \) = (7.5 ± 0.6) × 10−4 emu (g Oe)−1 and saturated value of the superparamagnetic magnetization at large field Ms(∞,T) = 0.240 ± 0.003 emu g−1, for T = 293 K. For the other isotherms, we find (assuming that Ns does not change with T)
where Ms(∞,T) = 0.323 ± 0.003, 0.385 ± 0.003 emu g−1, for T = 193 K, 120 K.
Equations (6), (7) allow to find the number Ns and the value μs(T) for our samples. The M(H) curves for 04PFT sample are well fitted by Eq. (2) with the parameter values, Ns = 6.3 × 1014, μs(T)/μB = 6.6 × 104, 5.5 × 104, 4.1 × 104 for T = 120, 193, 293 K, respectively.
Assuming that the superspin moments display a mean-field like temperature dependence \( \mu_{\text{s}} (T) = \mu_{\text{s}} (0)\sqrt {1 - T/T_{\text{c}} } \), we obtain μs(0)/μB ≈ 7.9 × 104, Tc ≈ 400 K.
For x = 0.5, \( \left. {\frac{{\partial M_{s} \left( {H,293} \right)}}{\partial H}} \right|_{H = 0} \) = 1.5 × 10−4 emu (g Oe)−1, Ms(∞,T) = 0.089, 0.072, 0.058 emu g−1 for T = 120, 193, 293 K, respectively. It gives Ns= 1.8 · 1014, μs(T)/μB= 4.8 × 104, 4.3 × 104, 3.5 × 104 for T = 120, 193, 293 K, μs(0)/μB≈ 5.5 × 104, Tc ≈ 490 K.
Let us consider the possibility to explain the superparamagnetism of our samples by magnetic impurity phases. In Refs. [50, 51], the hysteresis loops in PFT and solid solutions PFT–PbTiO3 were supposed to be due to the presence of lead hexaferrite (magnetoplumbite) PbFe12O19. At room temperature, the saturation magnetization of the hexaferrite is ~ 50 emu g−1 [60, 61], or 0.88μB per Fe spin. Thus, ns12 = 4.7 × 104 of Fe spins in a particle of PbFe12O19 would have a magnetic moment μs(T)/μB = 4.1 × 104 at T = 293 K that would explain the superparamagnetism of 04PFT sample (see fourth row of Table 2). The size of the particle would be ~ 110 Å, because the volume of PbFe12O19 unit cell is 695 Å3 [62] and it contains 2 formula units, i.e., 24 Fe ions. Then the volume fraction of PbFe12O19 in our sample would be 0.7% and the mass fraction—0.48%; it can not be detected by XRD. But, as we have mentioned in the “Discussion” section, such a small fraction of the magnetic impurity phase can not produce appreciable ME response for two-phase composite material [53]. Also, against the presence of this impurity in our samples is the fact that the Curie temperature of bulk PbFe12O19 is 720 K [60, 63], which can be reduced in nanoparticles, but seems to be still much larger than Tc= 400–490 K found by us. If nonmagnetic ions substitute for Fe in the hexaferrite, the Curie temperature and the saturation magnetization are reduced dramatically [64,65,66,67]. In order to explain the superparamagnetism in our samples by such a diluted magnetoplumbite impurity, we would have to assume a larger fraction of the impurity phase, which is not observed by XRD.
In Ref. [52], the pyrochlore Pb3FeTaO7 impurity was supposed to be responsible for room-temperature hysteresis in PFT–PT solid solutions. Pb3FeTaO7 has 8 formula units in the cubic unit cell with a = 10.5 Å. The saturation magnetization at room temperature is ~ 2.7 emu g−1. We obtain ns,pyr = 8.8 · 104 of Fe spins in a particle. The particle size is 233 Å, the volume fraction is ~ 7%, and the mass fraction is 9%. But our samples have no more than 2.4% of the pyrochlore admixture.
Therefore, neither PbFe12O19 nor Pb3FeTaO7 can be responsible for the magnetism and magnetoelectricity of the considered materials.
Magnetoelectric coupling
Within a phenomenological Landau–Ginzburg–Devonshire approach, linear and biquadratic ME couplings contribution to the system free energy are described by the terms μijPiMj and ξijklPiPjMkMl, respectively (P is polarization and M is magnetization, and μij and ξijkl are corresponding tensors of linear and biquadratic ME effects, respectively) [10, 19, 68]. In ceramics, the elements of the tensors are averaged and we are dealing with scalar LGD equations. If we start the description from the symmetric paraelectric paramagnetic phase, we should retain only biquadratic ME coupling
where ξMP is the coupling parameter that is assumed to weakly depend on temperature. The terms FP and FM depend only on polarization and magnetization, respectively. For the second-order phase transitions they have the usual form in SI units system
where only αP,M have strong temperature dependence α ~ (T – TC). For small external fields, we may write the solutions of Eq. (8) in the form
where Ps, Ms are spontaneous polarization and magnetization that become nonzero in the ordered phases; χE,M are dielectric and magnetic susceptibilities. Then ME coupling has the form
that allows us to express various magnetoelectric coefficients via ξMP
Due to the superparamagnetic contribution, the value of β will increase with temperature lowering according to the temperature dependence of magnetic susceptibility as [χM(T)]2 ~ 1/T2.
Using the value of β(T) obtained from experiments, we can estimate the biquadratic ME coefficient ξMP from the equation
We got ξMP ≈ –2.9 × 10−5 J m3 A−2 C−2 for the solid solution with x = 0.4 by substituting the room temperature values of parameters: β ≈ 0.15 × 10−15 s A−1, the spontaneous electric polarization Ps≈3.0 μC cm−2, susceptibilities dielectric χFE ≈2000, and magnetic (mass) χM ≈0.63 emu (g kOe)−1. For x = 0.5, we have obtained ξMP≈ − 0.0014 J m3 A−2 C−2, using β ≈ 0.54 × 10−15 s A−1, Ps≈ 4.4 μC cm−2, χFE ≈2000, χM ≈0.142 emu (g kOe)−1. All parameters were determined from the data of our experiments reported in previous sections.
Let us note that Eq. (11) is appropriate for the description of the paramagnetic phase in small fields where magnetization is proportional to H. For the description of the superparamagnetic phase at higher fields, we should consider another expression for the free energy part FM
where \( Z = {{\sinh \left[ {\frac{{2\left( {S + 1} \right)\mu_{\text{s}} \left( T \right)H}}{{2Sk_{\text{B}} T}}} \right]} \mathord{\left/ {\vphantom {{\sinh \left[ {\frac{{2\left( {S + 1} \right)\mu_{\text{s}} \left( T \right)H}}{{2Sk_{\text{B}} T}}} \right]} {\sinh \left[ {\frac{{\mu_{\text{s}} \left( T \right)H}}{{2Sk_{\text{B}} T}}} \right]}}} \right. \kern-0pt} {\sinh \left[ {\frac{{\mu_{\text{s}} \left( T \right)H}}{{2Sk_{\text{B}} T}}} \right]}} \) is the partition function for an isolated spin S in the magnetic field H. For S ≫ 1 this gives \( M_{\text{s}} \left( {H,T} \right) = - {{\partial F_{\text{M}} } \mathord{\left/ {\vphantom {{\partial F_{\text{M}} } {\partial H}}} \right. \kern-0pt} {\partial H}} \), Eq. (2). We will neglect the contribution from paramagnetic phase. Then instead of Eq. (14) we have
and
In the experiment, the ME effect is manifested as a polarization Pac induced by a small ac magnetic field hac under application of dc field Hdc. With using collinear dc and ac magnetic fields H = Hdc + hac sin ωt, we obtain a generalization of Eq. (4)
Hence, the generalization of Eq. (5) shows that the amplitude of magnetoelectric current IME is proportional to dM2/dH (see inset in Fig. 9a).
The above consideration can only provide a qualitative agreement with the experiment. A more realistic theory should take into account at least a distribution of moment values.
Rights and permissions
About this article

Cite this article
Glinchuk, M.D., Kuzian, R.O., Zagorodniy, Y.O. et al. Room-temperature ferroelectricity, superparamagnetism and large magnetoelectricity of solid solution PbFe1/2Ta1/2O3 with (PbMg1/3Nb2/3O3)0.7(PbTiO3)0.3. J Mater Sci 55, 1399–1413 (2020). https://doi.org/10.1007/s10853-019-04158-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-04158-4