
Abstract
Two sets of the carbon materials containing nitrogen and cobalt were prepared by simultaneous carbonization of CH3CN and decomposition of Co–Al layered double hydroxides (LDHs) (of Co/Al atomic ratios equal to 2 or 3) at 600, 700 and 800 °C followed by treatment with HCl. The physicochemical features of the samples were characterized by means of XRD, TG, EA, XPS, XRF, FT-IR, SEM, Raman spectroscopy and nitrogen sorption. All of the composites were active in oxygen reduction reaction (ORR) in alkaline medium. Their ORR behaviour was determined by the contribution of structural defects and N-doping in graphitic lattice. The beneficial effect of metallic cobalt encapsulated inside the N-CNTs on the ORR performance was also taken into consideration. The studies showed that the likelihood of the existence of Co–N–C species and their participation in ORR seemed to be suppressed by the formation of CoAl2O4. The samples prepared at 600 and 700 °C appeared to be more active in ORR than that obtained at 800 °C. The number of electrons involved in ORR (2.89, 2.82 and 2.77) decreased with increasing synthesis temperature for the samples prepared with LDHs of Co/Al = 2 as a consequence of lowering concentration of nitrogen (3.2, 2.4 and 1.5 wt%), in particular diminishing contribution of pyridinic nitrogen, and decreasing exposure of graphitic edges (ID1/IG ~ 2.6, 2.0 and 1.0). The ORR performance of the samples prepared with LDHs of Co/Al = 3 is comparable to that of the corresponding samples prepared with LDHs of Co/Al = 2.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Structural defects in graphitic lattice play a beneficial role in the ORR activity of the carbon materials as O2 adsorption is preferred on the edge graphene plane rather than on the basal graphene plane [1]. Thus, the edge planes show the ORR activity, while basal planes remain virtually inactive in ORR. Qu reported that the catalytic activity of activated carbon materials for ORR in alkaline solution was determined by the contribution of the edge plane on the surface.
Jiang et al. [2] prepared a series of carbon materials by carbonization of benzene at different temperatures (700, 800 and 900 °C) in the presence of MgO, which had various specific surface areas (i.e. 1713, 1009 and 614 m2g−1) and showed different ID/IG (the area ratios of two bands D- and G-bands), i.e. 2.29, 1.99 and 1.69. The samples were examined as ORR electrocatalysts in alkaline solution. The authors demonstrated that the intrinsic carbon defects significantly contributed to their ORR activity. They experimentally and computationally confirmed that two typical carbon defects, i.e. pentagon and zigzag edge, were the ORR active sites.
Defective sites in the carbon framework are formed when nitrogen doping takes place. N-doping of the carbon lattice increases the presence of graphene edge by facilitating the formation of pentagonal defects in hexagonally arranged carbon atoms, causing disruption to the graphene structure, and by introducing irregularities to the curvature in the graphene stacking, which lead to an enhanced ORR activity [3,4,5].
N-doped carbon materials possess electrocatalytic activity towards oxygen reduction reaction (ORR). The total nitrogen content does not dictate the ORR performance. The location of the nitrogen in carbon lattice plays an important role in the ORR activity. The incorporation of nitrogen into the carbon framework may result in pyridinic (denoted as N-6), pyrollic (denoted as N-5) and quaternary (denoted as N-4) nitrogens, which are claimed to be responsible for the ORR activity of the N-doped carbon materials [6,7,8]. While the pyridinic nitrogen and pyrrolic nitrogen always have nitrogen located on the graphitic edge, quaternary nitrogen can be both “edge-N” and “bulk-like-N”. The edge-N quaternary assignment is bonded to three carbons: one interior carbon and two edge carbons [9].
It is not well defined, which type of N-containing groups are related to 2-electron and which ones to 4-electron electroreduction of oxygen reaction [10]. The molecular oxygen adsorbed on the carbon atom adjacent to nitrogen atom by the side-one mode (known as the Yeager model) facilitates a desired 4-electron reduction reaction of oxygen leading finally to water molecule [11]. Some authors [12,13,14] claim that N-5 and N-6 are responsible for 4-electron reduction reaction, whereas N-4 is responsible for 2-electron reduction reaction.
Due to the presence of a lone pair of electrons, nitrogen has tendency to form complexes with transition metals such as cobalt or iron. Transition metal ions bonded to nitrogen on the surface of the N-doped carbon materials have been reported to be active in ORR [15,16,17,18,19].
In addition, transition metal used in synthesis of the N-doped carbon materials may influence the type of N-containing groups incorporated in the carbon framework [20], which results in further enhancement of the ORR activity. Oh and Kim [21] claimed that cobalt favored the formation of N-4 and N-6 and suppressed the formation of N-5, whereas an opposite opinion prevails in other works, i.e. that cobalt favored the formation of N-5 species [6]. Therefore, carbon materials doped with N, Co or Fe are intensively studied as the electrocatalysts for ORR, to replace commonly used, expensive Pt-based catalyst systems [15, 16, 21,22,23].
Among various organic compounds used as carbon (C) and nitrogen (N) source, acetonitrile (CH3CN) is a convenient one utilized in chemical vapour deposition method for synthesis of the N-doped carbon materials.
Using CH3CN as C/N source for the synthesis of the ORR electrocatalysts began by Dodelet et al. [24, 25] almost two decades ago, in the late nineties. They obtained active and stable electrocatalysts by pyrolysis of CH3CN at 1000 °C in the presence of iron deposited on carbon black (using various Fe loadings, i.e. 2 and 10 wt%). Various N-containing groups such as N-6, N-5, N-4 and nitrogen bonded to oxygen (denoted as N–O) were detected in those N-doped carbon materials. The sample derived from 2 wt% Fe/C had no carbon nanotubes (CNTs), whereas that derived from 10 wt% Fe/C contained CNTs. The sample derived from 2 wt% Fe/C contained iron in oxidized forms, whereas that derived from 10 wt% Fe/C contained iron mainly in carbidic and metallic forms. There was no evidence of the presence of Fe–Nx bonds.
Alexeyeva et al. [26] obtained the electrocatalyst by pyrolysis of CH3CN at 800 °C in the presence of Co/Al2O3. The studied sample containing N-CNTs appeared to be more active in ORR carried out in alkaline than in acidic medium.
Kundu et al. [27] compared the ORR performance of the electrocatalysts containing N-CNTs prepared via the carbonization of CH3CN in the presence of Co/SiO2 at lower (550 °C) and higher (750 °C) temperatures. Before electrochemical measurements, the as-prepared samples were treated with acid. The activity and selectivity of the samples in ORR in the acidic solution decreased with increasing synthesis temperature. The sample prepared at lower temperature had higher degree of structural defects (the intensity ratio of two bands D- and G-bands, ID/IG ~ 1.3) than that prepared at higher temperature (ID/IG ~ 1.2). The authors concluded that Co nanoparticles being covered with carbonaceous layers were not accessible by the electrolyte and they were not catalytically active. The presence of Co–N–C species was also excluded. It was shown that the catalytic activity of the samples was determined by exposure of graphitic edge plane.
A lot of work has been done to develop the ORR electrocatalysts by Ozkan et al. [28,29,30,31,32,33,34]. They obtained the catalysts by pyrolysis of CH3CN in the presence of either Fe- or Co-containing precursors deposited on various supports such as carbon black (C), alumina (Al2O3), magnesia (MgO) and silica (SiO2). Before testing in electrocatalysis, the as-prepared catalysts were additionally treated with acid and/or base. It appeared that the acid-treated catalysts were more active in ORR in the acidic solution than the corresponding as-prepared ones, as acid treatment improved the access to the active sites. In some cases, the washed samples showed improved ORR selectivity. They also pointed out that the samples prepared in the presence of transition metal-containing supports were much more active than those obtained with pure supports, in particular SiO2 and MgO. This indicated that the addition of transition metals (Fe, Co) to the supports would be recommended for developing ORR electrocatalysts.
The most active and stable catalysts were prepared by a 2-h pyrolysis of CH3CN at 900 °C in the presence of the supports impregnated with 2 wt% Fe. As a result, the Fe-containing species were encased in the final N-doped carbon nanostructures. The authors suggested that activity/selectivity of the electrocatalysts in ORR might be correlated with the extent of graphitic edge plane exposure.
From the studies performed in Ozkan’s group [30, 31, 34], it was unclear if the pyridinic nitrogen had a direct impact on the ORR activity or if it was just a marker for edge plane exposure, as it was located on the edge plane and the most active catalysts had both the highest exposure of graphitic edge plane and the highest content of pyridinic nitrogen.
Biddinger et al. [9] examined the role of graphitic edge plane exposure in carbon nanostructures for ORR using two types of carbon nanofibers: one with high edge plane exposure and one with minimal edge plane exposure. It was found that carbon nanostructure on its own was not a factor for improved ORR activity. They showed that carbon nanostructures with high edge plane exposure, like stacked platelets, provided the appropriate locations for nitrogen to be incorporated into the graphitic matrix. This observation was consistent with the previous findings that the edge orientation is more preferable for the functional groups to become incorporated into the graphitic lattice than the basal orientation; thus, higher concentration of the edge orientation would result in larger coverage of the functional groups [1].
According to the above-mentioned literature, the use of Al2O3 as a support for transition metals seems to be beneficial as it allowed preparing the N-doped carbon materials, which had relatively high specific surface areas and high nitrogen contents. In addition, the carbonaceous materials formed in the presence of Al2O3 by carbonization of acetonitrile are characterized by relatively high content of graphitic edge planes, which promote the ORR activity.
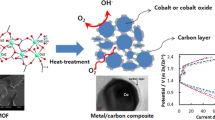
Our pervious works [35, 36] as well as the results reported by others [37, 38] showed that various double layered hydroxides (LDHs), among them Co–Al systems, were the effective substrates allowing the synthesis of N-doped carbon materials active in ORR. During thermal treatment upon CVD procedure with CH3CN, Co–Al LDHs were converted into a mixture of metal oxides, among them cobalt oxides were partially reduced to metallic cobalt. The presence of metallic cobalt nanoparticles promoted the formation of carbon nanostructures. Xue et al. [39] synthesized N-doped CNTs by carbonization of a mixture of methane and acetonitrile at 500 °C with the use of calcined Co–Al LDHs. They observed that the contribution of pyridinic nitrogen in the carbon lattice increased with increasing CH3CN flow. Thus, CH3CN could contribute more pyridinic nitrogen doping into the CNT lattice.
Various Co/Al atomic ratios (equal to 2 or 3) in Co–Al LDHs were used, as the concentration of cobalt during synthesis of N-doped carbon materials determined the content of nitrogen in the obtained samples [40].
The studies devoted to the samples derived from CH3CN and Mg–Co–Al LDHs showed an advantageous effect of acid treatment procedure resulting in partial elimination of inorganic component from the as-prepared materials, similarly to what was observed by other authors [30, 32, 33]. The acid-treated samples derived from CH3CN and Mg–Co–Al LDHs were more active/selective in ORR than the as-prepared ones because inorganic component blocked to high extent the access of oxygen to the active sites [36]. However, there are also reports on the negative impact of acid treatment on the ORR performance of washed catalysts [32, 41].
In this work, we report on the carbon materials doped with nitrogen and cobalt derived from CH3CN and Co–Al LDHs, prepared according to the procedure previously described [35], but additionally subjected to the aqueous solution of HCl. So, the fabricated materials are called acid-treated samples in contrast to as-prepared ones [35].
The as-prepared materials (derived from CH3CN and LDHs of Co/Al = 2) consisted of 27–35 wt% carbonaceous component and about twice higher content (65–73 wt%) of inorganic component (Co, CoO, Al2O3 and CoAl2O4) [35]. In the present work, inorganic component is partially removed from the as-prepared samples by dissolution in the aqueous solution of HCl. Although the inorganic component is still present in the acid-treated samples, its content is much below that of carbonaceous one (64–72 wt%). The proportion of these two components is rather similar in two series of the acid-treated materials.
The aim of this research is to study the relationship between physicochemical and electrochemical properties of the acid-treated composites with synthesis conditions. The electroreduction of oxygen, one of the most extensively studied electrochemical reactions, was chosen as a test reaction of the electrocatalytic properties of the prepared composites. These data should be useful/utilized for the rational design of electrocatalysts/electrode materials.
We also examine the studied samples as supports for Pd particles. The incorporation of nitrogen into the carbon framework creates structural defects, which can serve as adsorption centres for deposited Pd particles [42, 43]. Thus, the presence of N-containing groups in the carbon framework may influence the interaction between Pd particles and carbon support.
The activity, selectivity and stability of newly prepared N-doped carbon materials were also compared with commercial catalyst, i.e. 20 wt% Pt/C.
Materials and methods
Materials
Cobalt (II) nitrate hexahydrate [Co(NO3)2·6H2O, 99.0%, for analysis] was purchased from ACROS ORGANICS, whereas aluminium nitrate nonahydrate [Al(NO3)3·9H2O, min 98.8%, pure p.a.], sodium hydroxide (NaOH, min 98.8%, pure p.a.), potassium hydroxide (KOH, min 85%, pure p.a.), hydrogen peroxide (H2O2, min 29%, pure p.a.), acetonitrile (CH3CN, min 99.9%, for HPLC) and acetone [(CH3)2CO, min 99.5%, pure p.a.] were purchased from POCH S.A. All of the chemicals were used as received. Alumina powder for polishing (particle size of 0.3 μm) was purchased from Buehler. Nafion® perfluorinated ion-exchange resin was purchased from Sigma-Aldrich. Argon (Ar, 99.999%) and oxygen (O2, 99.95%) were purchased from Linde Gas. Water was purified using a Milli-Q purification system (Millipore).
Preparation of the N-doped carbon materials
The samples of LDHs (with Co/Al atomic ratios of 2 or 3) used as the precursors of both cobalt and alumina species were synthesized via method outlined in our previous publication [35].
Figure S1 shows the XRD patterns of two materials precipitated in the aqueous solutions of various Co/Al atomic ratios, equal to 2 or 3. Only one crystalline phase, exhibiting a layered structure like in LDHs, is detected by XRD in both materials. The XRD patterns of Co–Al LDHs show four characteristic reflections corresponding to (003), (006), (012) and (110) diffractions planes [44]. The Co/Al atomic ratios in the obtained samples determined by ICP (2.4 and 3.5) are consistent with the nominal Co/Al atomic ratios (2 and 3) used in the starting aqueous solutions.
The preparation of the N-doped carbon materials involved simultaneous decomposition of Co–Al LDHs and carbonization of CH3CN at 600, 700 and 800 °C. The sample of LDHs (0.5 g) (in an alumina boat) placed in a horizontal tube furnace was heated under a flow of Ar (30 mL min−1) to the required temperature (600, 700 and 800 °C). Then, the Ar stream saturated with CH3CN vapour was introduced and the sample was maintained at the target temperature for 30 min, and slowly cooled down under pure Ar stream.
Total amount of carbonaceous material in the as-prepared samples was evaluated from TG measurements performed in air up to 1000 °C. A single weight loss event occurring between 270 and 590 °C can be ascribed to the burning of amorphous/graphitic carbons [35].
In order to remove inorganic moiety from the as-prepared samples, they were treated with hydrochloric acid for 24 h using two different procedures. The samples derived from LDHs of Co/Al = 2 were agitated ultrasonically for 60 min using less concentrated hydrochloric acid solution (25.7 wt% HCl). The samples derived from LDHs of Co/Al = 3 were ultrasonically agitated for 30 min using more concentrated hydrochloric acid solution (36.5 wt% HCl). Finally, the resulting samples were separated, washed thoroughly with water and dried in a dry box at 50 °C.
The acid-treated samples prepared with LDHs of Co/Al = 2 are further denoted as C-2 series, whereas those synthesized at 600, 700 and 800 °C are further labelled as C-600-2, C-700-2 and C-800-2.
The acid-treated samples prepared with LDHs of Co/Al = 3 are further denoted as C-3 series, whereas those synthesized at 600, 700 and 800 °C are further labelled as C-600-3, C-700-3 and C-800-3.
Characterization of the physicochemical properties of the N-doped carbon materials
Inductively coupled plasma optical emission spectroscopy (ICP-OES) was applied to measure cobalt and aluminium concentrations.
Thermogravimetric (TG) analysis was performed using a STA 409PC (Netzsch) thermal analyser up to 1000 °C under static air condition with a heating rate of 5 °C min−1.
Elemental analysis (EA) was performed using an element analyser (vario EL III) to determine carbon, hydrogen and nitrogen contents.
X-ray fluorescence (XRF) analysis was carried out with EDX 3600 H XRD spectrometer from Skyray Instrument equipped with X-ray source (40 kV, 220 μA).
Powder X-ray diffraction (XRD) analysis was performed using a PANalytical X’Pert PRO powder diffractometer with Cu Kα radiation (40 kV, 30 mA). The XRD patterns were recorded in the range of 5°–139° (2θ) with a step size of 0.05°.
Raman spectroscopic measurements were performed by means of a Renishaw InVia spectrometer equipped with Ar ion laser (514 nm, power 50 mW), a Leica DMLM confocal microscope and a CCD detector. The Raman spectra were recorded using 5% of the power, in the spectral range of 200–3500 cm−1 with a resolution of 3 cm−1. Nine scans were accumulated to ensure a sufficient signal-to-noise ratio.
Fourier transform infrared spectroscopic (FT-IR) measurements were performed by means of Nicolet 380 FT-IR spectrometer. A mixture of the sample (1 mg) and KBr (200 mg) was pressed to form the transparent disc used in spectroscopic measurements. The FT-IR spectra (400–4000 cm−1) were obtained by co-addition of 64 scans recorded with a resolution of 4 cm−1. Spectra processing was performed using Thermo Scientific OMNIC™ software package.
X-ray photoelectron spectroscopic (XPS) measurements were conducted using a spectrometer equipped with Al Kα X-ray source (1486 eV, 12 kV, 20 mA) and a hemispherical analyser (R4000, Gammadata Scienta). XPS analysis provides general information on surface elemental composition up to 10 nm in the surface layer thickness.
Nitrogen adsorption/desorption analysis was carried out at − 196 °C using an Autosorb-1 (Quantachrome) surface area and the pore size analyser. Prior to volumetric sorption measurement, the samples were preheated and degassed under vacuum at 150 °C for 18 h. Specific surface area (SBET) was calculated using BET (Brunauer–Emmett–Teller) method based on adsorption data in the partial pressure of 0.1 < p/p0 < 0.35. Total pore volume (Vtot) was determined by the amount of nitrogen adsorbed at p/p0 = 0.99. Micropore volume (Vmicro) was estimated by the t-method, whereas mesopore volume (Vmeso) was calculated by using the data from the adsorption branch of nitrogen isotherm and the BJH (Barrett–Joyner–Halenda) method. The model of slit-like pore geometry was used in the studies to extract pore size distribution information.
Scanning electron microscopy (SEM) images were recorded using a JEOL JSM-7500F field emission apparatus (with an operating voltage of 15 keV). Energy dispersive spectroscopy (EDS) analysis of SEM images was performed. The depth of analysis by EDS is up to 1 μm. The samples were analysed without any surface treatment. The elemental composition of the samples by EDS was performed on the surface area of the samples equal to (40 µm × 50 µm) 2000 µm2.
Characterization of the electrochemical properties of the N-doped carbon materials
Electrochemical measurements were performed by means of the potentiostat/galvanostat PGSTAT204 (Metrohm Autolab) using an electrochemical cell in conventional three-electrode configuration. A Pt wire and an Ag/AgCl/KClsat electrode were used as a counter and reference electrodes, respectively. Glassy carbon (GC) disc (5 mm in diameter) was used as a substrate for electrochemical measurements. A GC substrate was polished in the aqueous alumina suspension on a polishing cloth, and then cleaned by ultrasonic treatment in Milli-Q water and acetone. The sample of the studied materials (2.5 mg) was ultrasonically mixed with 1 wt% Nafion suspension in aliphatic alcohols (a mixture of ethanol and n-propanol) (250 μL) for 20 min. Then, the resulting suspension (5 μL) was spread on a GC disc and dried in air at room temperature. The loading of the studied materials on GC was 0.25 mg cm−2. Cyclic voltammograms (CVs) were recorded at potential sweep rates of 5, 10, 20, 50, 100 mV s−1 in the aqueous solution of 0.1 M KOH. Linear polarization measurements were conducted at a sweep rate of 5 mV s−1 using rotating disc electrode (RDE) with rotation rate from 400 to 1600 rpm in alkaline medium, which was either saturated with oxygen or bubbled with argon. The temperature of 0.1 M KOH (25 ± 1 °C) was maintained by JULABO (ED-5) thermostat. All of the potentials are referred to Ag/AgCl/KClsat.
Results and discussion
Evaluation of the physicochemical properties
The as-prepared samples of C-2 and C-3 series were subjected to acid treatment in order to eliminate inorganic compounds, formed during CVD experiments through thermal decomposition of Co–Al LDHs. The weight loss after acid treatment was higher for the samples prepared at 600 °C (ca. 55 wt%) than for the samples obtained at 700 and 800 °C (ca. 45 wt%).
TG measurements reveal that inorganic residue still remains in the acid-treated samples. As shown in Table 1, the inorganic residue corresponds to ca. 28–36 wt% in C-2 series, while its content is somewhat lower (ca. 21–28 wt%) in C-3 series. The difference in the content of the inorganic residue for C-2 and C-3 series is rather related to various contents of carbonaceous deposit in the samples before acid treatment than to the effectiveness of the removal of inorganic compounds via dissolution in acid. The as-prepared samples derived from LDHs of Co/Al = 2 contain less carbonaceous deposit (27–35 wt%) than those derived from LDHs of Co/Al = 3 (32–42 wt%). As a consequence, the acid-treated samples of C-2 series have lower content (64–72 wt%) of carbonaceous component than those of C-3 series (72–79 wt%). Therefore, C-2 series have higher content of the inorganic residue than C-3 series.
The content of inorganic component is substantially reduced compared to that in the as-prepared samples. The carbonaceous component predominates in all of the acid-treated samples, as its contribution corresponds to ca. 64–79 wt%. The elemental analysis indicates that besides carbon and hydrogen, nitrogen is also present in all of the acid-treated samples.
The data obtained by XRF analysis (Table 2) indicate that the content of cobalt in the acid-treated samples (4–10 wt%) is significantly reduced in comparison with the initial samples (27–35 wt%). XRF analysis reveals also that more cobalt remains in the acid-treated samples of C-2 series than in their counterparts of C-3 series. It is also noticed that the content of cobalt varies with the synthesis temperature being the highest, i.e. 9.7 and 7.0 wt%, in the samples prepared at the highest temperature (800 °C) (Table 2).
The XRD analysis (Fig. S2a, b) confirms that the acid-treated samples are composed of carbonaceous component, such as graphite-like carbon (the reflections at 2θ of 26.3° and 43.1°) and two Co-containing crystalline phases, such as metallic cobalt (Co) (the reflections at 2θ of 44.3°, 51.7°, 76.1° and 92.6°) and cobalt aluminium oxide (CoAl2O4) (the shoulders at 2θ of 32.0°, 37.9°, 46.0°, 60.9° and 67.1°) [45, 46].
The dominating carbonaceous component of the samples seems to be more turbostratic than crystalline (graphitic). The disorder of graphite-like structure may be caused by the presence of nitrogen atoms in the carbon framework. The total content of nitrogen, determined by elemental analysis (Table 1), is below 4 wt% and decreases with rising synthesis temperature (600, 700 and 800 °C). On the other hand, Co/Al atomic ratio in Co–Al LDHs does not affect the content of nitrogen in the samples prepared at the same temperature. For example, the N/C atomic ratios calculated by EA and XPS are close for C-700-2 and C-700-3 (Tables 1, 3).
Partial graphitization of the carbonaceous component can be deduced from the C/H atomic ratio (Table 1). In a single hydrogen-terminated graphene ribbon (one ring wide and infinite in length), the C/H atomic ratio equal to 2 would be expected. This C/H atomic ratio increases with growing contribution of graphitic carbon. All of the studied samples show the C/H atomic ratios above 2, and the C/H atomic ratios of both C-2 and C-3 series remarkably increase with rising synthesis temperature (600, 700 and 800 °C). The C/H atomic ratios are almost twice higher for C-3 series compared to their counterparts of C-2 series. It shows that the higher Co/Al atomic ratio in LDHs, the more graphitic carbon is formed in the obtained N-doped carbon materials. This observation is consistent with the results of the Rietveld analysis data (Table 4), indicating that the content of graphitic carbon is higher for C-3 series (ca. 84–93 wt%) than for C-2 series (ca. 72–83 wt%).
Similarly to elemental analysis and XRD, Raman spectroscopy also reveals that the degree of graphitization increases with rising carbonization temperature from 600 up to 800 °C for C-2 (Fig. 1) and C-3 series. Two strong bands centred at about 1360 and 1590 cm−1 observed in the spectra of all of the samples are commonly considered to be a combination of four Lorentzian-shaped bands (G, D1, D2 and D4) at about 1580, 1350, 1620 and 1200 cm−1, respectively, and a Gaussian-shaped band (D3) at 1500 cm−1 [47]. These bands can be attributed to an ideal graphitic lattice (G), graphene layer edges (D1), graphene layer surface (D2), amorphous carbon (D3) and other types of disordered graphitic lattice (D4). The area ratios of various components (D1, D2, D3, D4 and G) of the Raman spectra are presented in Table 5. The analysis of the Raman spectra of both C-2 and C-3 series indicates reduced number of structural defects (ID1/(IG + ID1 + ID2)) and reduced contribution of amorphous carbon (ID3/IG) in the carbonaceous materials formed at higher temperature. Moreover, higher temperature leads to the thicker graphitic domains (ID2/IG). The contribution of amorphous carbon is identical for the samples prepared at the same temperature, irrespective of different Co/Al atomic ratios in LDHs used in CVD experiments.

The Raman spectra of C-2 series
The Raman spectra of C-2 series show also two very weak bands centred at 2710 and 2930 cm−1 (Fig. 1). The former can be attributed to the first overtone of the D1 band, whereas the latter can be assigned to a combination of the G and D modes characteristic of disturbed graphitic structures [47]. According to literature [4], N-doping may suppress the intensity of the band at ~ 2700 cm−1. This effect is also observed in the spectra of our samples with larger amounts of nitrogen, i.e. prepared at 600 and 700 °C (Fig. 1).
The XPS results confirm that carbon dominates on the surface of C-2 series as its concentration reaches the values of 68.7, 69.6 and 49.9 wt% (Table 3). The C 1s core level spectra of C-600-2, C-700-2 and C-800-2 are displayed in Fig. 2a. All of the C 1s signals consist of six peak components. The main peak component at binding energy of 284–285 eV can be assigned to the sp2–hybridized graphitic carbon (C=C) [5, 11]. Its contribution to the total area of the C 1s peak is similar (45.8, 44.0 and 47.4%) for C-600-2, C-700-2 and C-800-2, indicating that, irrespective of the synthesis temperature, approximately 45% of the surface carbon detected by XPS is represented by the sp2-hybridized graphitic units.

The XPS spectra of a series of C-2 showing a C 1s, b N 1s, c O 1s and d Co 2p3/2 signals
Our previous studies showed that treating the as-prepared samples with acid insufficiently eliminated non-carbonaceous species, namely Co- and/or Al-containing compounds formed through the thermal decomposition of Co–Al LDHs [35]. Similarly, metal particles (Fe, Co, Ni) and metal oxides (SiO2 and MgO) wrapped with carbonaceous deposit were observed by Matter et al. [28, 31] in the acid-treated carbon materials studied as the electrocatalysts for ORR.
Thermal decomposition of Co–Al LDHs results in the formation of Co3O4 and Al2O3. During CVD experiments, Co3O4 can be reduced by carbon (via CoO) to metallic cobalt. On the other hand, Co3O4 together with Al2O3 can be converted into spinel (CoAl2O4). Both products, i.e. metallic cobalt and spinel, are formed at 600, 700 and 800 °C.
The XRD diffraction patterns (Fig. S2a, b) of the acid-treated samples show the sets of reflections characteristic of crystalline Co- and/or Al-containing phases, such as metallic cobalt and cobalt aluminium oxide. The presence of amorphous phases, involving Co- and/or Al-containing species, cannot be excluded. The Rietveld analysis of the XRD patterns allows calculating the contribution of crystalline phases (expressed by wt%) in the samples before and after acid treatment (Table 4).
The as-prepared samples derived from LDHs of Co/Al = 2 contain larger amounts of metallic cobalt than their counterparts derived from LDHs of Co/Al = 3. Perhaps, better dispersion of Co3O4 on Al2O3 during the CVD experiments carried out with LDHs of Co/Al = 2 than in the case of LDHs of Co/Al = 3 favours the formation of metallic cobalt. It is worth to underline that there is no significant difference in the content of metallic cobalt in the as-prepared materials obtained at 600 and 700 °C, whereas the content of metallic cobalt is distinctively higher for the samples prepared at 800 °C.
The fraction of metallic cobalt (expressed by Cometallic/Cototal atomic ratio) in the initial samples is relatively high and equal to ~ 0.8, ~ 0.9 and ~ 1.0 for those prepared at 600, 700 and 800 °C, respectively, irrespective of Co/Al atomic ratio in Co–Al LDHs. It demonstrates that as the CVD temperature increases the concentration of metallic cobalt increases at the expense of that of cobalt oxide (CoO).
The fraction of metallic cobalt drastically decreases in the acid-treated materials showing that large amounts of metallic cobalt, most probably those not being covered with carbonaceous layers, are removed during acid treatment.
It is likely that in the initial samples prepared at lower temperatures (600 and 700 °C) metallic cobalt particles may be more loosely intermixed with the carbonaceous component than in those prepared at higher temperature (800 °C). Therefore, metallic cobalt particles, in particular those not being clingy covered with carbonaceous layers, can be more easily removed via acid treatment.
The amounts of remained metallic cobalt in the acid-treated samples grow with increasing synthesis temperature, being the highest, i.e. 10.8 and 6.4 wt%, in the samples prepared at the highest temperature (800 °C). In addition, the contribution of metallic cobalt is higher for C-2 series than C-3 series, especially for the samples prepared at 700 and 800 °C. For the samples prepared at 600 °C, i.e. C-600-2 and C-600-3, the contribution of metallic cobalt is similar.
The content of crystalline CoAl2O4 in the acid-treated materials is almost twice higher for C-2 series (ca. 14–20 wt%) than for C-3 series (ca. 7–10 wt%). It seems that the content of spinel in the final acid-treated samples depends on the Co/Al atomic ratio in LDHs. Apparently, better distribution of Co3O4 among Al2O3 during the CVD experiments carried out with LDHs of Co/Al = 2 than in the case of LDHs of Co/Al = 3 favours the formation of spinel.
Knowing that the conversion of Co3O4 and Al2O3 into CoAl2O4 depends on the concentration of oxidized form of cobalt, i.e. Co3O4, one may expect that the content of spinel should vary in an opposite way to the content of metallic cobalt. As mentioned above, the contribution of metallic cobalt in the as-prepared samples increases with increasing CVD temperature and, additionally, it is higher in the samples derived from LDHs of Co/Al = 2 than in those derived from LDHs of Co/Al = 3. Since the reflections characteristic of spinel are not seen in the XRD patterns of the as-prepared samples such scenario cannot be directly proofed by XRD. Furthermore, the XRD patterns of the acid-treated samples also do not support strictly this hypothesis because the amounts of spinel are higher for C-2 series than for C-3 series.
The formation of spinel during CVD experiments carried out with CH3CN and Co–Al LDHs is certainly disturbed by the formation of metallic cobalt.
We assume that the application of LDHs of Co/Al = 2 results in large quantity of spinel in the as-prepared samples. Moreover, the amounts of spinel decrease with increasing synthesis temperature, while the amounts of metallic cobalt increase. It is well known that spinel is difficult to dissolve in acid [35, 36]. Thus, large amounts of spinel in the as-prepared samples derived from LDHs of Co/Al = 2 lead to relatively large amounts of spinel in the acid-treated samples (C-2 series).
The above-mentioned assumption can be supported by the fact that the highest contribution of spinel (19.7 wt%) in C-2 series is observed for the sample prepared at the lowest temperature (600 °C).
CVD temperature influences not only the quantity of spinel but also its crystalline structure. The ability of spinel to dissolve in acid depends on its crystallinity. Smaller spinel crystallites, formed at lower temperature, can more easily dissolve in acid than larger ones, formed at higher temperature.
We assume that the application of LDHs of Co/Al = 3 results in small quantity of spinel in the as-prepared samples; therefore, its quality (crystallinity) plays an important role in acid treatment. It can be illustrated by the fact that the lowest contribution of spinel (6.6 wt%) in C-3 series is observed for the sample obtained at the lowest temperature (600 °C), whereas the highest contribution of spinel (9.9 wt%) is observed for the sample obtained at the highest temperature (800 °C). Such trend is not noticed for C-2 series as in the case of C-2 series the quantity of spinel (in the as-prepared samples) plays more crucial role in acid treatment than its crystallinity.
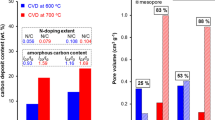
Our previous work and other reports showed that removal of inorganic compounds (metal/metal oxides) from the as-prepared samples via treating with acid uncovers the entrance to the pores present in the carbonaceous component [28, 36, 48]. This effect can be also observed for the acid-treated samples of C-2 series, which have approximately twice higher specific surface area than the corresponding as-prepared samples (Table 2). The nitrogen adsorption/desorption isotherms can be classified as type IV [49], which implies the presence of mesopores.
Specific surface area (SBET) of both C-2 and C-3 series (Table 2) decreases with increasing synthesis temperature (600, 700 and 800 °C). The samples prepared at the lowest temperature (600 °C) have the highest SBET, while the samples prepared at the highest temperature (800 °C) have the lowest SBET. The trend towards lowering SBET with increasing carbonization temperature can be related to the formation of the carbon material with improved structural ordering revealed also by EA, XRD and Raman spectroscopy.
SEM micrographs displayed in Fig. 3a–c illustrate that C-2 series contain carbon tubular nanostructures with spaghetti-like appearance. Carbon nanotubes (CNTs) were also observed for the corresponding as-prepared samples [35]. It is evident that the observed CNTs are formed on metallic cobalt nanoparticles acting as the catalysts for their growth. According to Rietveld analysis, the average diameter of metallic cobalt crystallites is calculated to be 11 and 19 nm for C-700-2 and C-800-2, respectively. The diameter of CNTs ranges roughly from 10 to 100 nm and slightly increases with rising CVD temperature, which could be related to larger metallic cobalt particles in the samples prepared at 800 °C.

The SEM images of a series of C-2: C-600-2 (a), C-700-2 (b), C-800-2 (c)
Figures S3a, b, S4a, b and S5a, b illustrate that the Co-containing species are evenly distributed throughout the carbonaceous component. The lower concentrations of cobalt obtained by XPS (4.2, 1.4 and 1.9 wt%) than those determined by EDS (5.3, 3.8 and 7.1 wt%) for C-2 series suggest that the Co-containing species are covered with a relatively thick carbonaceous layers, i.e. the shells of CNTs. Considering the difference in the analysis depth between XPS (up to 10 nm) and EDS (up to 1 μm), it implies that the Co-containing species, being involved in the growth of carbon nanostructures, is encapsulated into CNTs. Similar observations are reported for other carbon nanostructures synthesized in the presence of the Co-containing precursors [16, 50, 51].
Besides CNTs, the studied samples contain also carbonaceous grains showing other morphologies such as large aggregates of oriented platelets. In addition, EDS analysis (Figs. S4a, b, S5a, b) reveals that some large, irregular aggregates are enriched with the Al- and O-containing species, the products of thermal decomposition of Co–Al LDHs, for example amorphous Al2O3. They are difficult to dissolve in acid, especially when formed at high temperature; therefore, their population is the highest in the samples prepared at the highest temperature (800 °C) (Fig. 3c, f).
Much higher concentrations of Al obtained by XPS (9.7, 14.0 and 24.7 wt%) than those determined by EDS (6.0, 6.2 and 7.7 wt%) for C-2 series can be related to the presence of the Al-containing species in the form of relatively big particles, as illustrated by SEM images (Figs. S3a, b, S4a, b).
In conclusion, the main (carbonaceous) component of the acid-treated samples (64–79 wt%) possesses graphitic ordering but also contain structural defects (showing edge and basal orientations on the surface). Some amorphous carbon is also present in all of the samples, and its content is lower in the samples prepared at higher temperature. The carbonaceous component is composed of CNTs and plate-like carbon particles, being doped with nitrogen. The concentration of nitrogen, i.e. below 4 wt%, decreases with increasing synthesis temperature. Its content is similar for the samples prepared at the same temperature irrespective of Co/Al atomic ratio in Co–Al LDHs. The inorganic component of the samples is difficult to separate from carbonaceous component via acid treatment. All of the samples contain both metal and metal oxides (21–36 wt%) well intermixed with carbonaceous component. The inorganic compounds are mostly covered with carbonaceous layers. The Co-containing species, such as metallic cobalt particles, are rather located inside the CNTs, whereas other compounds, such as CoAl2O4 and Al2O3, seem to be rather wrapped with clingy carbonaceous sheets, outside the CNTs. The contributions of metallic cobalt and CoAl2O4 vary with various synthesis temperatures and depend on the Co/Al atomic ratio in Co–Al LDHs.
Evaluation of the electrochemical properties
The oxygen reduction reaction (ORR) in alkaline medium was chosen to characterize the electrocatalytic properties of the studied N-doped carbon materials. The electrochemical behaviour was assessed by linear sweep voltammetry (LSV) measurements using rotating disc electrode (RDE). In addition, cyclic voltammetry (CV) technique was applied to examine hydrogen peroxide reduction reaction.
Figures S6a–c and S7a–c show the LSV curves recorded in O2-saturated 0.1 M KOH at a scan rate of 5 mV s−1 with rotation rate changing from 400 to 1600 rpm for GC coated with C-2 and C-3 series, respectively.
The comparison of RDE results recorded for C-2 series at the rotation rate of 1600 rpm (Fig. 4) demonstrates a gradual decrease in the ORR activity with increasing synthesis temperature, from 600 to 800 °C (Table 6). Similar trend in the ORR activity is observed for C-3 series. It indicates that the synthesis temperature determines the ORR performance and the samples prepared at 600 °C are the most active.

The current–potential relations recorded in the aqueous solution of O2-saturated 0.1 M KOH by sweeping the potential at 5 mV s−1 for a GC coated with C-2 series
Similar influence of the synthesis temperature on the onset potential was observed in our previous work on the N-doped carbon materials also derived from CH3CN but prepared with the use of Mg–Co–Al LDHs as inorganic precursors [36].
The comparison of RDE results (Fig. S8) reveals slightly different ORR performance of the samples prepared at the same temperature but with LDHs of Co/Al = 2 and Co/Al = 3. It demonstrates that the samples obtained with LDHs of Co/Al = 2 are more active than those obtained with LDHs of Co/Al = 3. The results for the samples prepared at 700 °C were omitted from the comparison for the sake of clarity. RDE results for the samples prepared at 700 °C are situated between the polarization curves obtained for the samples prepared at 600 and 800 °C.
The kinetic currents (obtained between − 0.13 and − 0.15 V) illustrate that the samples of C-2 series are more active than their counterparts of C-3 series (Fig. S9).
The specific surface area (determined by nitrogen sorption) of C-2 series differs by a factor of about 1.1–1.2 compared to their counterparts of C-3 series (Table 2), and their kinetic current also differs by a factor of about 1.1–1.5. Thus, higher specific surface area could be a possible reason for explaining the observed enhanced electrochemical activity of C-2 series compared to C-3 series.
Judging by the value of kinetic current, the samples of both C-2 and C-3 series prepared at 600 °C (C-600-2 and C-600-3) are the most active for ORR, whereas those prepared at 800 °C (C-800-2 and C-800-3) are the least active.
Tafel plots of mass-transport-corrected currents obtained for the studied samples using the data from the corresponding polarization curves (Figs. S6a–c and S7a–c) are shown in Fig. S10a, b, and the corresponding Tafel slopes either in the low (at the potentials between − 0.10 and − 0.15 V) or in high current regions (at the potentials between − 0.15 and − 0.25 V) are collected in Table 6. The Tafel behaviour of C-2 and C-3 series is similar. Moreover, the obtained Tafel slopes are close to the values reported on various N-doped carbon materials studied for ORR in 0.1 M KOH [14, 36, 52].
It has been reported [2, 46, 53,54,55] that the structural defects present in the carbon materials promoted their activity for ORR. Similar effect is observed for the studied samples. The number of defects, i.e. a contribution of graphene layer edges (ID1/IG, Table 5), is higher in the samples prepared at lower temperature (600 °C), which are more active in ORR than those prepared at higher temperature (800 °C) (Fig. S8, Table 6).
Additionally, the electrocatalytic activity of the N-doped carbon materials may also be related to the surface N-containing groups [6,7,8]. Their presence in the studied samples is confirmed by XPS (Table 3). The N 1s core level spectra of C-2 series are shown in Fig. 2b. The N 1s signals consist of four peak components ascribed to pyridinic nitrogen (at 398–399 eV), pyrrolic nitrogen (at 400–401 eV), quaternary nitrogen (at 402–403 eV) and oxidized nitrogen (at 404–405 eV) [35]. All of these N-containing species are observed at various contributions except pyrrolic nitrogen, which content is similar (39.2–40.3%), irrespective of synthesis temperature. Pyrrolic nitrogen is the dominating N–C species in the samples prepared at higher temperature (700 and 800 °C), whereas pyridinic nitrogen dominates in the sample prepared at lower temperature (600 °C). When the synthesis temperature grows, the contribution of pyridinic nitrogen decreases, whereas that of quaternary nitrogen increases. The contribution of N–O is very low in all of the samples.
The observed enhanced ORR performance with decreasing synthesis temperature for C-2 series (Fig. 4) seems to correlate with growing contribution of pyridinic nitrogen. Judging by the value of the onset potential, the following order of the ORR activity is observed: C-600-2 > C-700-2 > C-800-2 in line with decreasing contribution of pyridinic nitrogen. This observation is also consistent with previous reports [27, 31, 34] on other N-doped carbon materials derived from CH3CN. Those studies showed that a coexistence of a large number of structural defects and dopants (N-species), in particular pyridinic nitrogen, promoted the ORR activity.
Apart from the N-containing groups also the O-containing groups being present in the studied samples may affect the ORR performance. XPS confirms the presence of oxygen atoms in the studied samples. The O 1s core level spectra for C-2 series are shown in Fig. 2c. The O 1s signals consist of five peak components such as oxygen atoms connected to metal atoms (at 530–531 eV), oxygen atoms in −OH groups (at ~ 532 eV), oxygen atoms in C=O and C–O–C groups (at 533–534 eV), oxygen atoms in carboxyl (COOH) and carbonate (CO32−) groups and in adsorbed water (H2O) (at 535–536 eV) and oxygen atoms in the oxygen molecules (O2) or bonded to aromatic carbon compounds (at ~ 537 eV) [11, 35]. The major part of oxygen (ca. 83–98%) is associated with oxygen atoms incorporated in the carbon framework, whereas the minor part of oxygen (ca. 2–17%) originates from the metal oxides, i.e. CoAl2O4, Al2O3.
According to literature [11, 56,57,58,59], carbonyl (including carbonyl oxygen of quinones) and carboxyl groups may enhance the ORR activity. It is claimed that the carbon atom located near the O-containing groups may be responsible for the adsorption of molecular oxygen by the end-one mode (known as the Pauling model), which facilitates 2-electron oxygen reduction reaction leading to HO2− [11]. In addition, the O-containing groups may be involved in ORR as mediators [57, 60].
More precise analysis of the XPS spectra of C-2 series reveals that the total contribution of carbonyl and carboxyl groups decreases with rising synthesis temperature from 600 up to 800 °C, which is in line with the observed activity order, showing that the sample synthesised at 600 °C is more active than that prepared at 800 °C.
It should be noted that besides previously discussed N- and O-species, also Co–N–C species could participate in both oxygen and hydrogen peroxide reduction reactions [49, 58]. According to literature [15, 16, 18, 21], the Co–N–C bonds are stable up to 700 °C. However, other papers report the existence of the Co–N–C bonds in the samples prepared at higher temperatures such as 800 or 900 °C [61, 62].
The presence of the Co–N–C bonds may be reflected in the FT-IR spectra by the band at low wavenumbers, i.e. ca. 418 cm−1 [62], while in XPS Co 2p core level spectra by the Co 2p3/2 signal at binding energy of ca. 779–781 eV [62, 63].
In the 400–1000 cm−1 spectral region of all of the studied samples (Fig. S11a, b), the bands characteristic of CoAl2O4 are only observed [64, 65].
The surface elemental composition of the studied samples is analysed by XPS (Table 3). The highest total surface concentration of cobalt, i.e. 4.2 wt%, is observed for C-600-2, and it is almost twice higher than in C-700-2 and C-800-2 (1.4 and 1.9 wt%, respectively).
Figure 2d demonstrates that the Co 2p3/2 signal for C-2 series consists of four peak components, which we assigned to cobalt carbide (CoxC) (at ~ 778 eV), metallic cobalt (Co) (at ~ 781 eV), cobalt in oxidized form, i.e. CoAl2O4, (at ~ 782 eV). The presence of crystalline Co and CoAl2O4 is proved by XRD (Fig. S2a, b). The last peak (at ~ 785 eV) can correspond to the cobalt ions in an electronegative surrounding, i.e. Co–Cl. Chlorine is observed for all of the studied materials (Table 3).
The surface content of metallic cobalt (estimated by XPS) in C-2 series decreases with increasing synthesis temperature, i.e. 0.16, 0.08 and 0.05 at% for the samples prepared at 600, 700 and 800 °C, respectively. When the synthesis temperature grows Co particles agglomerate and become larger as revealed by XRD, i.e. the average diameter of metallic cobalt crystallites is 11 and 19 nm for C-700-2 and C-800-2, respectively. As a consequence, Co particles are less distributed in the carbonaceous component. EDS maps (Figs. S3b, S4b, S5b) show that the population of the Co-containing species is lower for C-700-2 and C-800-2 than for C-600-2.
On the other hand, larger metallic cobalt becomes coated with thicker graphitic layers. As mentioned above, the average outer diameter of CNTs in the studied samples increases with increasing synthesis temperature [35]. Based on literature [39], this effect may be a result of larger Co particles and therefore higher number of walls in the CNT shell.
Based on the XPS spectra showing Co 2p3/2 signal, cobalt also occurs in carbidic form. Its surface content in the sample prepared at 600 °C (0.22 at%) is comparable to that in the sample prepared at 800 °C (0.22 at%). The complementary information, provided by XPS spectra showing C 1s signal, demonstrates that the content of metal–carbon species decreases with increasing synthesis temperature, i.e. 3.11, 0.72 and 1.60 at% for the samples prepared at 600, 700 and 800 °C, respectively.
The surface content of CoAl2O4 (estimated by XPS) in C-2 series decreases with increasing synthesis temperature, i.e. 0.38, 0.10 and 0.10 at% for the samples prepared at 600, 700 and 800 °C, respectively. This effect is consistent with decreasing bulk contribution of crystalline CoAl2O4 (determined by XRD). Furthermore, the crystallinity of CoAl2O4 somewhat improves with increasing synthesis temperature and as a consequence CoAl2O4 is less distributed throughout the carbonaceous component. Beside this, the thickness of the graphitic layers resided on CoAl2O4 grows with increasing synthesis temperature. As a consequence, CoAl2O4 becomes more shielded.
Metallic cobalt exposed on the surface of the electrode materials should manifest in the CV curves recorded in alkaline medium by a pair of peaks associated with redox transformation between cobalt hydroxide (Co(OH)2) and cobalt oxyhydroxide (CoOOH) [66]. Such electrochemical response is not observed for any of the studied samples. The existing metallic cobalt is not accessible by the electrolyte because its particles, as mentioned above, are enclosed inside the carbon nanotubes.
It has already been reported that coating of metallic cobalt with the carbonaceous layers makes it inactive in ORR owing to the lack of accessibility for the oxygen molecules [27, 67,68,69]. However, an opposite opinion prevails in other works [70, 71], i.e. metallic iron encapsulated inside the N-CNTs may affect the electronic properties of the outer wall of N-CNT shell, where O2 is adsorbed and reduced.
Deng et al. [70] experimentally and computationally demonstrated that the interaction of Fe nanoparticles encapsulated inside the CNTs with the graphitic walls of the CNTs might lead to a decreased local work function on the carbon surface. In addition, doping N into the carbon lattice might further reduce the local work function. The reduced local work function at the carbon surface with iron sitting below was expected to enhance its ORR activity.
The peak components (characteristic of cobalt carbide and metallic cobalt) at ca. 778 and ca. 781 eV in the Co 3p3/2 core level spectra (Fig. 2d), are located at binding energy higher by 0.4–0.6 and 0.2–1 eV, respectively, in C-600-2 in comparison with those in C-700-2 and C-800-2. Similar tendency is observed for the peak component (characteristic of metal–carbon species) at ca. 283 eV in the C 1s core level spectra (Fig. 2a), which is located at binding energy higher by 0.4–0.8 eV in C-600-2 in comparison with that in C-700-2 and C-800-2. The observed trends may be due to the interaction of Co nanoparticles and carbonaceous layers, which is especially pronounced in the sample prepared at 600 °C containing small Co nanoparticles wrapped with thin and highly N-doped graphitic layers. Among the samples of C-2 series, this sample is the most active in ORR.
Zhang et al. [37] obtained active and selective electrocatalyst from melamine (containing 67 wt% of nitrogen) and Co–Al LDHs (Co/Al = 2). Its electrocatalytic performance for ORR (n = 3.7) has been related to the N-doped CNTs and the encapsulated Co nanoparticles. The sample contained also amorphous Al2O3, whereas no cobalt occurred in the form of CoAl2O4.
In the case of our samples derived from acetonitrile and Co–Al LDHs, besides Co encased in the N-CNTs and Al2O3, also reasonable amounts of CoAl2O4 are present. It shows that under applied synthesis conditions (using acetonitrile as carbon/nitrogen source), Co3O4 not only can be reduced by carbon to metallic cobalt but also it can be converted together with Al2O3 into CoAl2O4. Similarly, high content of CoAl2O4 was reported for the samples prepared via CVD experiments carried out with the use of Co–Al LDHs and acetonitrile vapour and/or other gaseous substrates like acetylene, methane [39, 48, 72].
On the other hand, the use of Co–Al LDHs and melamine, i.e. two solid materials during the carbonization process, seems to inhibit the formation of CoAl2O4. The Co-containing species being engaged to form CoAl2O4 may be less available for other ongoing processes. Thus, the reaction between Co3O4 and Al2O3, leading to the formation of CoAl2O4 may suppress the probability of the formation of Co–N–C species during CVD with the use of CH3CN and Co–Al LDHs. As a consequence, the studied samples may not contain Co–N–C species.
Among the N- and O-containing species, the ORR performance is controlled rather by the N- than O-containing groups. So we postulate that the N–C species observed for the studied samples (Fig. 2b) are involved in ORR.
In order to determine the number of electrons transferred in ORR in the presence of the studied samples, the RDE data in the limiting current regime are analysed [73]:
where i is the measured limiting current, n is the number of electrons transferred per oxygen molecule, F is the Faraday’s constant (96485 C mol−1), A is the geometric electrode area (0.20 cm2), D and C are the diffusion coefficient of dissolved oxygen (1.9 × 10−5 cm2 s−1) and the concentration of dissolved oxygen (1.2 × 10−6 mol cm−3) in 0.1 M KOH, respectively; υ is the kinematic viscosity of the electrolyte solution (0.01 cm2 s−1), and ω is the rotation rate (rad s−1) [74].
Figure 5 shows the Levich plots (i vs ω1/2) for C-2 series, which represent a relationship between the measured limiting current (at the potential of − 0.6 V) and the square root of the rotation rate. The plots obtained from experimental data are between the theoretical lines characteristic of 2- and 4-electron reaction pathways. Similar results are observed for C-3 series. It implies that besides the desired 4-electron oxygen reaction also the 2-electron oxygen reduction reaction contributed to some extent to the overall ORR over the investigated samples. Similar number of electrons, equal to 3, involved in ORR in alkaline medium was reported for the N-doped carbon materials, e.g. N-doped CNTs, N-doped CNFs and N-doped graphene [6, 7, 12, 53].

The Levich plots for oxygen reduction reaction over a GC coated with C-2 series denoted as C-600-2 (circles), C-700-2 (squares) and C-800-2 (triangles)
The number of electrons, i.e. 2.89, 2.82 and 2.77 (Table 6), slightly decreases with increasing synthesis temperature for C-2 series. The population of graphitic edge plane (ID1/IG, Table 5) and the concentration of N-containing groups (Table 3), in particular pyridinic nitrogen (Fig. 2b), for C-2 series decrease in the same order. The sample prepared at 600 °C showing the highest electron number (n = 2.89) has the highest concentration of graphitic edge plane sites (ID1/IG = 2.58) and the highest contribution of pyridinic nitrogen (42.3%). The sample prepared at 800 °C showing the lowest electron number (n = 2.77) has the lowest concentration of graphitic edge plane sites (ID1/IG = 1.02) and the lowest contribution of pyridinic nitrogen (31.6%). Thus, both surface features, i.e. the structural one represented by the defects in graphitic lattice and the chemical one represented by the N-containing groups participated in ORR.
The ability of both C-2 and C-3 series to reduce oxygen does not differ significantly (Table 6). The number of electrons involved in ORR is 2.82 and 2.71 for C-700-2 and C-700-3, respectively.
Figure S12 shows the specific capacitance for the samples prepared at 600 and 800 °C using LDHs of various Co/Al atomic ratios (2 or 3). The specific capacitance was calculated by dividing the capacitance by the specific surface area (determined by nitrogen sorption) (Table 2). The capacitance was calculated from the CV data (the voltammograms recoded in the aqueous solution of Ar-bubbled 0.1 M KOH, between + 0.3 V and − 0.6 V versus Ag/AgCl/KClsat, with various potential sweep rates of 10, 20, 50 and 100 mV s−1) using the current at the potential of − 0.15 V. Figure S12 illustrates that the samples of C-2 and C-3 series prepared at the same temperature have similar electrochemical accessibility of the pores.
Since ORR takes place on the external surface of the studied samples, our experimental results suggest that (a) not all of the active sites (revealed by XPS, Raman) are available for the oxygen molecules, (b) the active sites in the catalysts have ability to reduce oxygen by 2 and 4 electrons, but 2-electron ORR seems to be dominating.
In order to check the electrocatalytic performance of the studied samples towards hydrogen peroxide reduction, the CV curves were recorded in the aqueous solution of 0.1 M KOH with the addition of H2O2 (1.2 mM H2O2), between + 0.3 V and − 0.6 V versus Ag/AgCl/KClsat, with a potential sweep rate of 10 mV s−1. The reduction in hydrogen peroxide for C-2 series occurs at the potentials of − 0.14, − 0.17 and − 0.19 V for C-600-2, C-700-2 and C-800-2, respectively. Similar results, i.e. − 0.16, − 0.18 and − 0.22 V, are obtained for the samples prepared with LDHs of Co/Al = 3. It shows that the catalytic activity of both C-2 and C-3 series towards hydrogen peroxide reduction decreases with increasing temperature from 600 up to 800 °C. This trend in the activity is similar to that observed for ORR. In addition, judging by the value of the potential at the maximum of the peak reduction, the samples of C-2 series are more active than their counterparts of C-3 series. As mentioned before for ORR, C-2 series seems to have larger effective area (larger SBET) and therefore lower current density and lower overpotential of the reaction than C-3 series.
The comparison of the electrocatalytic performance of C-2 and C-3 series with the literature data for other electrocatalysts, containing carbon materials doped with N and Co, is shown in Table S1. It is difficult to make a clear comparison because the other authors used different Co, N, C precursors and synthesis conditions, which had impact on the surface properties of those catalysts and consequently on their ORR activity/selectivity. The samples derived from CH3CN and Co–Al LDHs studied in this work are less selective for 4-electron ORR than other reference materials.
However, the samples studied in this work, in particular those prepared at 700 and 800 °C, have the ORR performance comparable to that of N-free carbon materials prepared by carbonization of benzene in the presence of MgO [2]. The activity of the latter has been related to the presence of various structural defects in graphitic lattice, such as pentagon and zigzag edge defects. Our ORR results in terms of the onset potential and the number of electrons correspond closely to the reported data, i.e. − 0.09 and − 0.13 V, respectively, and n = 2.9.
The synthesis of the electrocatalysts with the application of acetonitrile and Co–Al LDHs does not lead to more active and selective ORR catalysts than those already reported in the literature. Nevertheless, electrochemical measurements show that the obtained N-doped carbon materials have the potential for the electrocatalytic production of hydrogen peroxide and/or for electrochemical sensing (H2O2 detection) as other mesoporous N-doped carbon materials [75,76,77].
For comparison, the commercial catalyst labelled as Pt/XC-72 (20 wt% Pt/C) is also evaluated in ORR (Fig. S13a). All of the studied N-doped carbon materials appear to be less active and selective in ORR than Pt/C (Figs. S14a, S15a, Table S2).
We apply one of the studied samples, namely C-700-3, as a support for Pd particles. Pd particles with the diameter of ca. 10 nm, prepared according the procedure previously described by Kosydar et al. [78], were deposited onto C-700-3. We also deposited Pd particles on N-free carbon material, commercial Vulcan XC-72. The concentrations of Pd (determined by XRF) in Pd/C-700-3 and Pd/XC-72 are comparable for both samples and equal to 23.9 and 21.7 wt%, respectively.
Spreading Pd particles on C-700-3 and XC-72 affects the porosity (Table S3) and the pore size distribution of the obtained catalysts (Pd/C-700-3, Pd/XC-72). Pd particles seem to reside on the outer surface of the supports, between individual carbonaceous grains. Furthermore, agglomerated Pd particles provide the interparticle voids, with the diameters in the mesopore range. In the case of Pd/XC-72, Pd particles by locating on individual carbonaceous grains partially block the entrance to the pores (micropores) within carbonaceous grains.
The electrochemical measurements (Figs. S14b–d, S15b–d, Table S2) reveal that Pd/C-700-3 is more active and more selective for 4-electron ORR than Pd-free C-700-3. Both Pd-containing catalysts show similar ORR performance despite different surface properties of the carbon supports (in terms of grain morphology, the presence/absence of N-containing groups). It seems that the ORR behaviour of Pd/C-700-3 and Pd/XC-72 is mainly owing to the Pd-containing species.
Figure S16 demonstrates that the kinetics of ORR over Pd-containing catalysts is similar to that of Pt/C. The Tafel slopes in low (at potentials between − 0.04 and − 0.10 V) and high (at potentials between − 0.10 and − 0.20 V) current regions are summarized in Table S2. Similar Tafel slopes were observed for Pd particles supported on N-doped graphene [79].
Figure S17 compares the kinetic currents (obtained between − 0.05 and − 0.10 V) for Pd-containing catalysts and Pt/C catalyst. Both Pd-containing catalysts show lower kinetic current than Pt/XC-72. Pd/C-700-3 appears to be as active as Pd/XC-72. It implies that the influence of the carbon support on the ORR performance under applied conditions is negligible. We cannot confirm synergistic effect between N- and Pd-containing species in terms of electrocatalysis.
The stability of the Pd-containing catalysts and the corresponding supports is evaluated by subjected them to recording a series of 50 LSV curves in the aqueous solution of O2-saturated 0.1 M KOH between the open circuit potential (~ 0 V) and − 0.7 V at 1600 rpm with a potential sweep rate of 5 mV s−1 (a testing period lasts ca. 7000 s).
The stability test reveals that the onset potential remains unaffected (Fig. S18a–d) and the Tafel slope remains virtually unchanged (the data are not shown). No significant changes upon the stability test can be observed in the voltammograms (recorded in the aqueous solution of O2-saturated 0.1 M KOH, between + 0.4 V and − 1.0 V versus Ag/AgCl/KClsat, with a potential sweep rate of 10 mV s−1), which indicates that the surface properties of the catalysts are mostly maintained.
However, the number of electrons involved in ORR becomes somewhat lower than before the stability test, which shows that the selectivity for 4-electron ORR slightly decreases. It may be caused by the slow modification of the surface of the catalysts [80, 81].
Conclusions
All of the studied samples are able to reduce oxygen in 0.1 M KOH by 2- and 4-electron processes. Although the samples obtained at 600, 700 and 800 °C have different physicochemical properties (e.g. various concentrations of defects and nitrogen, different contributions of N–C species), they do not differ significantly in the catalytic activity/selectivity towards ORR. In addition, there is no distinct difference in the electrochemical properties of the samples prepared at the same temperature but using LDHs of various Co/Al atomic ratios, equal 2 or 3. The application of precursor containing higher content of cobalt, i.e. LDHs of Co/Al = 3, does not result in more active electrocatalysts.
The structural defects and the surface functional groups, especially N-containing ones, in the carbonaceous component, play a role in ORR. However, the influence of metallic cobalt encapsulated inside the N-CNTs on the ORR performance of the acid-treated samples cannot be completely excluded.
The probability of the formation of Co–N–C species during CVD experiments with the use of CH3CN and Co–Al LDHs (with various Co/Al atomic ratios of 2 or 3) may be suppressed by the conversion of Co3O4 and Al2O3 into CoAl2O4.
The N-doped carbon materials appear to be less active, selective and stable in ORR than commercial Pt-containing catalyst (20 wt% Pt/C). It appears that the N-doped carbon material (prepared at 700 °C) derived from CH3CN and LDHs with Co/Al = 3 is as good support for Pd particles as commercial carbon material (Vulcan XC-72). The presence of N-containing groups in the carbon framework does not enhance ORR performance of deposited Pd particles.
References
Qu D (2007) Investigation of oxygen reduction on activated carbon electrodes in alkaline solution. Carbon 45:1296–1301
Jiang Y, Yang L, Sun T, Zhao J, Lyu Z, Zhuo O, Wang X, Wu Q, Ma J, Hu Z (2015) Significant contribution of intrinsic carbon defects to oxygen reduction activity. ACS Catal 5:6707–6712
Sjöström H, Stafström S, Boman M, Sundgren J-E (1995) Superhard and elastic carbon nitride thin films having fullerene like microstructure. Phys Rev Lett 75:1336–1339
Maldonado S, Stevenson KJ (2005) Influence of nitrogen doping on oxygen reduction electrocatalysis at carbon nanofiber electrodes. J Phys Chem B 109:4707–4716
Maldonado S, Morin S, Stevenson KJ (2006) Structure, composition, and chemical reactivity of carbon nanotubes by selective nitrogen doping. Carbon 44:1429–1437
Shin D, Jeong B, Mun BS, Jeon H, Shin H-J, Baik J, Lee J (2013) On the origin of electrocatalytic oxygen reduction reaction on electrospun nitrogen– carbon species. J Phys Chem C 117:11619–11624
Lai L, Potts JR, Zhan D, Wang L, Kok Poh Ch, Tang Ch, Gong H, Shen Z, Lin J, Ruoff RS (2012) Exploration of the active center structure of nitrogen-doped graphene-based catalysts for oxygen reduction reaction. Energy Environ Sci 5:7936–7942
Shi H, Shen Y, He F, Li Y, Li A, Liu S, Zhang Y (2014) Recent advances of doped carbon as non-precious catalysts for oxygen reduction reaction. J Mater Chem A 2:15704–15716
Biddinger EJ, Ozkan US (2010) Role of graphitic edge plane exposure in carbon nanostructures for oxygen reduction reaction. J Phys Chem C 114:15306–15314
Yang Z, Nie H, Chen X, Chen X, Huang S (2013) Recent progress in doped carbon nanomaterials as effective cathode catalysts for fuel cell oxygen reduction reaction. J Power Sources 236:238–249
Zhong R-S, Qin Y-H, Niu D-F, Tian J-W, Zhang X-S, Zhou X-G, Sun S-G, Yuan W-K (2013) Effect of carbon nanofiber surface functional groups on oxygen reduction in alkaline solution. J Power Sources 225:192–199
Li H, Kang W, Wang L, Yue Q, Xu S, Wang H, Liu J (2013) Synthesis of three-dimensional flowerlike nitrogen-doped carbons by a copyrolysis route and the effect of nitrogen species on the electrocatalytic activity in oxygen reduction reaction. Carbon 54:249–257
Wong WY, Daud WRW, Mohamad AB, Kadhum AAH, Majlan EH, Loh KS (2012) Nitrogen-containing carbon nanotubes as cathodic catalysts for proton exchange membrane fuel cells. Diam Relat Mater 22:12–22
Chen Z, Higgins D, Chen Z (2010) Nitrogen doped carbon nanotubes and their impact on the oxygen reduction reaction in fuel cells. Carbon 48:3057–3065
Faubert G, Lalande G, Côté R, Guay D, Dodelet JP, Weng LT, Bertrand P, Dénès G (1996) Heat-treated iron and cobalt tetraphenylporphyrins adsorbed on carbon black: physical characterization and catalytic properties of these materials for the reduction of oxygen in polymer electrolyte fuel cells. Electrochim Acta 41:1689–1701
Nallathambi V, Lee J-W, Kumaraguru SP, Wu G, Popov BN (2008) Development of high performance carbon composite catalyst for oxygen reduction reaction in PEM proton exchange membrane fuel cells. J Power Sources 183:34–42
Bouwkamp-Wijnoltz AL, Visscher W, van Veen JAR, Tang SC (1999) Electrochemical reduction of oxygen: an alternative method to prepare active CoN4 catalysts. Electrochim Acta 45:379–386
Li S, Zhang L, Liu H, Pan M, Zan L, Zhang J (2010) Heat-treated cobalt–tripyridyl triazine (Co–TPTZ) electrocatalysts for oxygen reduction reaction in acidic medium. Electrochim Acta 55:4403–4411
Kramm UI, Herrmann-Geppert I, Fiechter S, Zehl G, Zizak I, Dorbandt I, Schmeißer D, Bogdanoff P (2014) Effect of iron-carbide formation on the number of active sites in Fe–N–C catalysts for the oxygen reduction reaction in acidic media. J Mater Chem A 2:2663–2670
Zhang H-J, Kong H-Ch, Yuan X, Jiang Q-Z, Yang J, Ma Z-F (2012) Influence of metal precursors on the catalytic activity and structure of non-precious metal electrocatalysts for oxygen reduction reaction. Int J Hydrog Energy 37:13219–13226
Oh HS, Kim H (2012) The role of transition metals in non-precious nitrogen-modified carbon-based electrocatalysts for oxygen reduction reaction. J Power Sources 212:220–225
Bezerra CWB, Zhang L, Lee K, Liu H, Marques ALB, Marques EP, Wang H, Zhang J (2008) A review of Fe–N/C and Co–N/C catalysts for the oxygen reduction reaction. Electrochim Acta 53:4937–4951
Kramm UI, Herrmann-Geppert I, Behrends J, Lips K, Fiechter S, Bogdanoff P (2016) On an easy way to prepare metal– nitrogen doped carbon with exclusive presence of MeN4-type sites active for the ORR. J Am Chem Soc 138:635–640
Lalande G, Côté R, Guay D, Dodelet JP, Wenig LT, Bertrand P (1997) Is nitrogen important in the formulation of Fe-based catalysts for oxygen reduction in solid polymer fuel cells? Electrochim Acta 42:1379–1388
Faubert G, Côté R, Guay D, Dodelet JP, Dénès G, Poleunis C, Bertrand P (1998) Activation and characterization of Fe-based catalysts for the reduction of oxygen in polymer electrolyte fuel cells. Electrochim Acta 43:1969–1984
Alexeyeva N, Tammeveski K, Lopez-Cudero A, Solla-Gullón J, Feliu JM (2010) Electroreduction of oxygen on Pt nanoparticle/carbon nanotube nanocomposites in acid and alkaline solutions. Electrochim Acta 55:794–803
Kundu S, Nagaiah TC, Xia W, Wang Y, van Dommele S, Bitter JH, Santa M, Grundmeier G, Bron M, Schuhmann W, Muhler M (2009) Electrocatalytic activity and stability of nitrogen-containing carbon nanotubes in the oxygen reduction reaction. J Phys Chem C 113:14302–14310
Matter PH, Wang E, Ozkan US (2006) Preparation of nanostructured nitrogen-containing carbon catalysts for the oxygen reduction reaction from SiO2- and MgO-supported metal particles. J Catal 243:395–403
Matter PH, Wang E, Arias M, Biddinger EJ, Ozkan US (2006) Oxygen reduction reaction catalysts prepared from acetonitrile pyrolysis over alumina-supported metal particles. J Phys Chem B 110:18374–18384
Matter PH, Ozkan US (2006) Non-metal catalysts for dioxygen reduction in an acidic electrolyte. Catal Lett 109:115–123
Matter PH, Wang E, Arias M, Biddinger EJ, Ozkan US (2007) Oxygen reduction reaction activity and surface properties of nanostructured nitrogen-containing carbon. J Mol Catal A 264:73–81
Singh D, Tian J, Mamtani K, King J, Miller JT, Ozkan US (2014) A comparison of N-containing carbon nanostructures (CNx) and N-coordinated iron–carbon catalysts (FeNC) for the oxygen reduction reaction in acidic media. J Catal 317:30–43
Singh D, Soykal II, Tian J, von Deak D, King J, Miller JT, Ozkan US (2013) In situ characterization of the growth of CNx carbon nano-structures as oxygen reduction reaction catalysts. J Catal 304:100–111
Matter PH, Zhang L, Ozkan US (2006) The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction. J Catal 239:83–96
Pacuła A, Socha RP, Zimowska M, Ruggiero-Mikołajczyk M, Mucha D, Nowak P (2013) Application of as-synthesized Co–Al layered double hydroxides for the preparation of the electroactive composites containing N-doped carbon nanotubes. Appl Clay Sci 72:163–174
Pacuła A, Uosaki K, Socha RP, Bielańska E, Pietrzyk P, Zimowska M (2016) Nitrogen-doped carbon materials derived from acetonitrile and Mg–Co–Al layered double hydroxides as electrocatalysts for oxygen reduction reaction. Electrochim Acta 212:47–58
Zhang S, Zhang Y, Jiang W, Liu X, Xu S, Huo R, Zhang F, Hu J-S (2016) Co–N–CNTs derived from triple-role CoAl-layered double hydroxide as an efficient catalyst for oxygen reduction reaction. Carbon 107:162–170
Wang J, Li L, Chen X, Lu Y, Yang W, Duan X (2017) A Co–N/C hollow-sphere electrocatalyst derived from a metanilic CoAl layered double hydroxide fort he oxygen reduction reaction, and its active sites in various pH media. Nano Res 10:2508–2518
Xue R, Sun Z, Su L, Zhang X (2010) Large-scale synthesis of nitrogen-doped carbon nanotubes by chemical vapour deposition using a Co-based catalyst from layered double hydroxides. Catal Lett 135:312–320
Fu S, Zhu C, Li H, Du D, Lin Y (2015) One-step synthesis of cobalt and nitrogen co-doped carbon nanotubes and their catalytic activity for the oxygen reduction reaction. J Mater Chem A 3(12718):12722
Mamtani K, Singh D, Tian J, Millet MJ-M, Miller JT, Co AC, Ozkan US (2016) Evolution of N-coordinated iron–carbon (FeNC) catalysts and their oxygen reduction (ORR) performance in acidic media at various stages of catalyst synthesis: an attempt at benchmarking. Catal Lett 146:1749–1770
Arrigo R, Schuster ME, Xie Z, Yi Y, Wowsnick G, Sun LL, Hermann KE, Friedrich M, Kast P, Hävecker M, Knowp-Gericke A, Schlögl R (2015) Nature of the N–Pd interaction in nitrogen-doped carbon nanotubes catalysts. ACS Catal 5:2740–2753
Sing K, Zou Z, Wang D, Bien Tan, Wang J, Chen J, Li T (2016) Microporous organic polymers derived microporous carbon supported pd catalysts for oxygen reduction reaction; impact of framework and heteroatom. J Phys Chem C 120:2187–2197
Pacuła A, Nowak P, Socha RP, Ruggiero-Mikołajczyk M, Mucha D, Bielańska E (2013) Preparation and characterization of the electroactive composites containing nickel nanoparticles and carbon nanotubes. Electrochim Acta 90:563–572
Pacuła A, Ikeda K, Masuda T, Uosaki K (2012) Examination of the electroactive composites containing cobalt nanoclusters and nitrogen-doped nanostructured carbon as electrocatalysts for oxygen reduction reaction. J Power Sources 220:20–30
Wu Y, Zang J, Dong L, Zhang Y, Wang Y, Zhang Y (2016) High performance and bifunctional cobalt-embedded nitrogen doped carbon/nanodiamond electrocatalysts for oxygen reduction and oxygen evolution reactions in alkaline media. J Power Sources 305:64–71
Sadezky A, Muckenhuber H, Grothe H, Niessner R, Poschl U (2005) Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43:1731–1742
Li F, Tan Q, Evans DG, Duan X (2005) Synthesis of carbon nanotubes using a novel catalysts derived from hydrotalcite-like Co–Al layered double hydroxide precursor. Catal Lett 99:151–156
Rouquerol F, Rouquerol J, Sing K (1999) Adsorption by powders and porous solids, principles, methodology and applications. Academic Press, London
Wei G, Wainright JS, Savinell RF (2000) Catalytic activity for oxygen reduction reaction of catalysts consisting of carbon, nitrogen and cobalt. J New Mater Electrochem Syst 3:121–129
Wang Z, Xiao S, Zhu Z, Long X, Zheng X, Lu X, Yang S (2015) Cobalt-embedded nitrogen doped carbon nanotubes: a bifunctional catalyst for oxygen electrode reactions in a wide pH range. ACS Appl Mater Interfaces 7:4048–4055
Ma G, Jia R, Zhao J, Wang Z, Song C, Jia S, Zhu Z (2011) Nitrogen-doped hollow carbon nanoparticles with excellent oxygen reduction performances and their electrocatalytic kinetics. J Phys Chem C 115:25148–25154
Higgins D, Chen Z, Chen Z (2011) Nitrogen doped carbon nanotubes synthesized from aliphatic diamines for oxygen reduction reaction. Electrochim Acta 56:1570–1575
Zhong G, Wang H, Yu H, Peng F (2014) The effect of edge carbon of carbon nanotubes on the electrocatalytic performance of oxygen reduction reaction. Electrochem Commun 40:5–8
Yasuda S, Yu L, Kim J, Murakoshi K (2013) Selective nitrogen doping in graphene for oxygen reduction reactions. Chem Commun 49:9627–9629
Alexeyeva N, Shulga E, Kisand V, Kink I, Tammeveski K (2010) Electroreduction of oxygen on nitrogen-doped carbon nanotube modified glassy carbon electrodes in acid and alkaline solutions. J Electroanal Chem 648:169–175
Kruusenberg I, Mondal J, Matisen L, Sammelselg V, Tammeveski K (2013) Oxygen reduction on graphene-supported MN4 macrocycles in alkaline media. Electrochem Commun 33:18–22
Biddinger EJ, von Deak D, Ozkan US (2009) Nitrogen-containing carbon nanostructures as oxygen-reduction catalysts. Top Catal 52:1566–1574
Zhang M, Yan Y, Gong K, Mao L, Guo Z, Chen Y (2004) Electrostatic layer-by-layer assembled carbon nanotube multilayer film and its electrocatalytic activity for O2 reduction. Langmuir 20:8781–8785
Wu J, Zhang D, Wang Y, Wan Y, Hou B (2012) Catalytic activity of graphene–cobalt hydroxide composite for oxygen reduction reaction in alkaline media. J Power Sources 198:122–126
Liu ZX, Liu BH, Li ZP (2014) Effects of hydrothermal treatment on the catalytic activity of cobalt-doped catalysts for oxygen reduction reaction. Int J Hydrog Energy 39:5689–5695
Wang H, Bo X, Wang A, Guo L (2013) Cobalt doped nanoporous hollow carbon spheres as novel non-precious metal oxygen reduction electrocatalysts. Electrochem Commun 36:75–79
Masa J, Zhao A, Xia W, Muhler M, Schuhmann W (2014) Metal-free catalysts for oxygen reduction in alkaline electrolytes: influence of the presence of Co, Fe, Mn and Ni inclusions. Electrochim Acta 128:271–278
Chen Z, Shi E, Li W, Zheng Y, Zhong W (2002) Hydrothermal synthesis and optical property of nano-sized CoAl2O4 pigment. Mater Lett 55:281–284
Deraz NM (2013) Formation and characterization of cobalt aluminate nano-particles. Int J Electrochem Sci 8:4036–4046
Huang H, Wang Q, Wei Q, Huang Y (2015) Nitrogen doped mesoporous carbon derived from copolymer and supporting cobalt oxide for oxygen reduction reaction in alkaline media. Int J. Hydrog Energy 40:6072–6084
Subramanian NP, Li X, Nallathambi V, Kumaraguru SP, Colon-Mercado H, Wu G, Lee J-W, Popov BN (2009) Nitrogen-modified carbon-based catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J Power Sources 188:38–44
Niwa H, Kobayashi M, Horiba K, Harada Y, Oshima M, Terakura K, Ikeda T, Koshigoe Y, Ozaki J, Miyata S, Ueda S, Yamashita Y, Yoshikawa H, Kobayashi K (2011) X-ray photoemission spectroscopy analysis of N-containing carbon-based cathode catalysts for polymer electrolyte fuel cells. J Power Sources 196:1006–1011
Pacuła A, Nowak P, Makowski W, Socha RP (2015) The influence of layered double hydroxide composition on the morphology, porosity and capacitive properties of nitrogen-doped carbon materials prepared via chemical vapor deposition. Microporous Mesoporous Mater 201:1–9
Deng D, Yu L, Chen X, Wang G, Jin L, Pan X, Deng J, Sun G, Bao X (2013) Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction. Angew Chem Int Ed 52:371–375
Jiang W-J, Gu L, Li L, Zhang Y, Zhang X, Zhang L-J, Wang J-Q, Hu J-S, Wei Z, Wan L-J (2016) Understanding the high activity of Fe–N–C electrocatalysts in oxygen reduction: Fe/Fe3C nanoparticles boost the activity of Fe–Nx. J Am Chem Soc 138:3570–3578
Hima IH, Xiang X, Zhang L, Li F (2008) Novel carbon nanostructures of caterpillar-like fibre and interwoven spheres with excellent surface super-hydrophobicity produced by chemical vapor deposition. J Mater Chem 18:1245–1252
Bagotsky VS (2006) Fundamentals of electrochemistry. Wiley, Hoboken
Vikkisk M, Kruusenberg I, Joost U, Shulga E, Tammeveski K (2013) Electrocatalysis of oxygen reduction on nitrogen-containing multi-walled carbon nanotube modified glassy carbon electrodes. Electrochim Acta 87:709–716
Lee Y-H, Li F, Chang KH, Hu C-C, Ohsaka T (2012) Novel synthesis of N-doped porous carbons from collagen for electrocatalytic production of H2O2. Appl. Catal. B: Environ. 126:208–214
Mao X, Rutledge GC, Hatton TA (2014) Nanocarbon-based electrochemical systems for sensing, electrocatalysis, and energy storage. Nano Today 9:405–432
Perazzolo V, Durante C, Pilot R, Paduano A, Zheng J, Rizzi GA, Martucci A, Granozzi G, Gennaro A (2015) Nitrogen and sulfur doped mesoporous carbon as metal-free electrocatalysts for the in situ production of hydrogen peroxide. Carbon 65:949–963
Kosydar R, Góral M, Gurgul J, Drelinkiewicz A (2012) The effect of support properties in the preparation of Pd size-controlled catalysts by “water-in-oil” microemulsion method. Catal Commun 22:58–67
Jukk K, Kongi N, Matisen L, Kallio T, Kontturi K, Tammeveski K (2014) Electroreduction of oxygen on palladium nanoparticles supported on nitrogen-doped graphene nanosheets. Electrochim Acta 137:206–212
Shao Y, Yin G, Gao Y (2007) Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J Power Sources 171:558–566
Kabir S, Zadick A, Atanassov P, Dubau L, Chatenet M (2017) Stability of carbon-supported palladium nanoparticles in alkaline media: a case study of graphitized and more amorphous supports. Electrochem Commun 78:33–37
Acknowledgements
This research received funding from National Science Centre in Poland within decision number DEC-2013/11/B/ST5/01417. This work was also financially supported by National Science Centre in Poland within the research project 2699/B/T02/2009/36 for the years 2009–2012. Raman spectra were recorded by means of the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-12-023/08). Prof. Alicja Drelinkiewicz and Prof. Paweł Nowak both from Jerzy Haber Institute of Catalysis and Surface Chemistry Polish Academy of Sciences are gratefully acknowledged for fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pacuła, A., Socha, R.P., Pietrzyk, P. et al. Physicochemical and electrochemical properties of the carbon materials containing nitrogen and cobalt derived from acetonitrile and Co–Al layered double hydroxides. J Mater Sci 53, 11292–11314 (2018). https://doi.org/10.1007/s10853-018-2427-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-018-2427-9