
Abstract
Here, we present a design of core–shell structured carbon@MnO composite nanospheres and investigate its electrochemical performance as an anode material for lithium-ion batteries. The core–shell carbon@MnO composite nanospheres are obtained from the intermediate product of carbon@MnO2 nanospheres by coating a MnO2 layer over the surface of the mesoporous carbon cores, followed by thermal treatment in an inert atmosphere. The morphology and crystal phase of the obtained nanospheres are examined, and the electrochemical properties as a lithium-ion battery anode material are studied. The results demonstrate that the ordered mesoporous carbon@MnO electrode shows remarkable enhancements in lithium storage capacity, rate capability and cycling stability, delivering an average capacity of 572 mAh g−1 at 500 mA g−1 over 1000 charge/discharge cycles. The morphology and phase of the core–shell carbon@MnO electrode material after extended cycling are examined by transmission electron microscopy and X-ray diffraction, which indicate the nanocrystalline rather than amorphous property of the cycled electrode. As MnO is a conversion-type electrode material, the potential polarization of the carbon@MnO composite electrode is also investigated, which exhibits a unique evolution as cycling proceeds.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rechargeable lithium-ion batteries, as a main or backup power source for electronic systems from portable electronic devices to electric vehicles, have been used extensively in our daily life due to their advantages such as high volumetric and gravimetric energy density, low self-discharge rate and no memory effect. The conventional anode material for lithium-ion batteries, graphite, has been used for many years due to its high electrochemical stability during lithium-ion intercalation/deintercalation, high conductivity and lithium-ion diffusivity, low intercalation potential and low cost. However, with a relatively low theoretical capacity (372 mAh g−1), graphite will soon hardly meet the ever-growing energy consumption requirements of modern electronics. Therefore, the exploitation and development of novel alternative anode materials with high electrochemical performance for lithium-ion batteries makes great economic value.
Transition metal oxides (TMO) such as Co3O4 [1, 2], Fe3O4 [3,4,5], NiO [6,7,8],MnOx [9,10,11,12,13,14,15], CuO [16, 17], ZnO [18, 19], etc., have attracted much research attention and been viewed as promising anode materials for next-generation lithium-ion batteries due to their outstanding theoretical energy storage capacities [20]. However, based on phase conversion mechanisms, these TMOs generally face common drawbacks such as large volume expansion during the charge/discharge process, low electron conductivities, high lithium insertion potential and large potential polarizations, resulting in undesirable cycling stability, low rate capability and low battery voltage. These problems could be relieved to a certain degree via rational design of the structure and composition of the electrode materials. For example, it has been demonstrated that nanosized electrode materials are more favorable for electron transfer and lithium-ion diffusion due to the shortened pathways. In addition, porous structures are advantageous for battery performance as they can accommodate larger volume change during the electrochemical conversion/deconversion process and provide larger electrode–electrolyte contact area. Moreover, composite materials are also considered to be an effective solution for improving the battery performance. The combination of carbon and TMO materials is a promising technique for high-performance electrode materials because of the potential synergistic effects. TMOs could provide higher lithium storage capacity, while carbon materials such as carbon nanotube, graphene, graphite can increase the overall conductivity of the electrode materials and buffer the large volume expansion of the TMOs. Previous research results have demonstrated the effectiveness of the carbon/TMOs combination [21,22,23,24,25,26,27]. For example, Zhou et al. [28] prepared a well-organized flexible interleaved composite of graphene nanosheets decorated with Fe3O4 particles, which showed a remarkably improved reversible capacity (1026 mAh g−1 after 30 cycles at 35 mA g−1 and 580 mAh g−1 after 100 cycles at 700 mA g−1) and cyclic stability for lithium-ion batteries over the commercial Fe3O4 or bare Fe2O3 counterparts. MnO and N-doped carbon hybrid, synthesized by Xiao et al., manifested high capacity, excellent cyclability and superior rate capability over bare MnO material, delivering a lithium storage capacity of 1699 mAh g−1 at 0.5 A g−1 (after 170 discharge–charge cycles) and retaining a high reversible capacity of 907.8 mAh g−1 after 400 cycles at a current density up to 5 A g−1 [29].
Herein, we report the synthesis of a core–shell structured composite of ordered mesoporous carbon (OMC) nanospheres and a coating of MnO nanoparticles. The electrochemical properties as the negative electrodes for lithium-ion batteries were investigated. It is found the incorporation of a layer of crystallized MnO nanoparticles endows a great increase in lithium storage capacity compared with the bare OMC nanospheres, along with a good rate capability and long-term cycling life. The microstructure and the crystallinity of the electrode materials after extended cycling were also examined. Finally, the potential polarization of the electrodes and its evolution with cycling, remaining largely unexplored in previous literature, were studied.
Experimental
Synthesis of OMC, OMC@MnO2 and OMC@MnO nanospheres
Figure 1 illustrates the synthetic route for the OMC@MnO nanospheres. OMC nanospheres were synthesized through a soft-template method reported in a previous study [30], using phenolic resol as the carbon precursor and a commercial triblock poly(ethylene oxide)–poly(propylene oxide)-poly(ethylene oxide) (PEO–PPO–PEO) polymer (Pluronic F127) as the mesopore-forming agent, followed by a high-temperature carbonization process. To prepare OMC@MnO2 nanospheres, 100 mg of the OMC nanospheres was first dispersed in 100 ml of deionized (DI) water in an ultrasonic bath. Then, 344 mg of KMnO4 powder was added in the solution and the solution was heated to 70 °C under magnetic stirring. The reaction was allowed to proceed for 3 h. The OMC@MnO2 product was collected via centrifugation, washed with DI water for several times and dried in an electric oven overnight. The OMC@MnO nanospheres were obtained by treating the OMC@MnO2 nanospheres in a furnace tube under flowing Ar at 600 °C for 3 h at a heating rate of 5 °C min−1.

Schematic diagram of the synthetic route for OMC@MnO nanospheres
Material characterization
The morphology and nanostructures of the OMC, OMC@MnO2 and OMC@MnO nanospheres were characterized by a Hitachi S4800 electron scanning microscope and a JEOL 2100 cryo-electron transmission microscope. The phase and crystal structures of the materials were examined by a Siemens D5000 X-ray diffractometer using Cu Kα radiation (40 kV, 30 mA). The XRD patterns of the film electrodes on copper foil were measured using a Philips X’pert XRD instrument. Raman spectroscopy was carried out on a Nanophoton Raman-12 system using a 532 nm excitation wavelength. X-ray photoelectron spectroscopy analysis was conducted on a Kratos Axis Ultra XPS system. The weight ratio of carbon for the samples was obtained through thermal gravimetric analysis using a TA Q50 instrument.
Electrochemical characterizations
The electrochemical properties of the materials were investigated using 2032 coin-type cells. The working electrode was prepared by mixing 70% of the active material (OMC, OMC@MnO2 or OMC@MnO nanospheres), 20% of the carbon conductive additive (superP) and 10% of polymer binder (carboxyl methyl cellulose, CMC) to form a homogenous paste which was then coated on a copper foil, dried at 120 °C in a vacuum oven for 12 h and punched into circular disks at a diameter of 10 mm. Metallic lithium was used as the counter and reference electrode and 1 M LiPF6 in a mixture of ethylene carbonate and diethyl carbonate solution (1:1 by mass ratio) as the electrolyte. The half-cells were assembled in an Ar-filled glovebox with both moisture and oxygen concentrations below 1.0 ppm. The electrochemical properties of the electrode materials were studied via cyclic voltammetry and galvanostatic charge/discharge measurements over a voltage range from 0.01 to 3 V.
Result and discussions
Figure 2a, b shows the SEM images of the obtained OMC nanospheres, which reveals that these OMC nanospheres are 100–200 nm in size. Figure 2c shows the TEM image of the OMC nanospheres, clearly revealing the ordered mesopores in the nanospheres. The OMC nanospheres were then coated with a MnO2 layer through the reaction with KMnO4, giving rise to core–shell structured OMC@MnO2 nanospheres. The reaction of KMnO4 and carbon could be described using the following equation:
where 344 mg of KMnO4 gives rise to 65.8 wt% of MnO2 in the OMC@MnO2 composite. This result is very close to the result obtained from the TGA measurement (34.6 wt% of carbon and 65.4 wt% of MnO2). The SEM images of the OMC@MnO2 nanospheres are shown in Fig. 2d, e, revealing that the MnO2 nanoflakes are formed over the surface of the OMC nanospheres. Figure 2f shows the TEM image of the OMC@MnO2 nanospheres, which displays more clearly the OMC core and the MnO2 shell with an approximate thickness of 60 nm. The selected area electron diffraction (SEAD) pattern arising from the MnO2 layer is presented in the inset, indicating that the obtained MnO2 nanoflakes are amorphous. The OMC@MnO2 nanospheres were then thermally treated at 600 °C in Ar atmosphere, which are finally transformed to OMC@MnO. Figure 2g, h shows the SEM images of the OMC@MnO sample. As can be seen, there is an obvious morphology change after the thermal treatment. The MnO2 nanoflakes are converted to MnO nanoparticles with an average diameter around 53 nm, as also demonstrated by the TEM image (Fig. 2i). The inset in Fig. 2i shows the HRTEM image of one MnO nanoparticle displaying clear lattice fringe with a distance of 0.222 nm, which is well indexed to (200) plane of MnO.

SEM and TEM images of a–c OMC, d–f OMC@MnO2, g–i OMC@MnO nanoparticles. Inset in c shows the HRTEM image of OMC. Inset in f shows the SAED pattern of the MnO2 layer over the OMC nanospheres. Inset in i shows the HRTEM image of one MnO nanoparticle. Scale bar, 500 nm for a, d, g, 300 nm for b, e, 200 nm for c, f, h, 100 nm for i
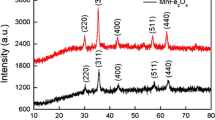
Figure 3a displays the XRD patterns of the OMC, OMC@MnO2 and OMC@MnO samples. No carbon diffraction peak is shown for the OMC nanospheres, indicating its amorphous property. The OMC@MnO2 also does not show any diffraction peak, which indicates the deposited MnO2 nanoflakes are also amorphous. After treated at 600 °C, strong diffraction peaks readily indexed to MnO (JCPDS No. 75-1090) are observed, indicating that the MnO2 has been reduced to MnO in inert atmosphere with the presence of carbon. The size of MnO nanospheres estimated from the XRD results is 49 nm, which is very close to the SEM observation. The Raman spectra of the OMC, OMC@MnO2 and OMC@MnO samples are shown in Fig. 3b. All the samples show broad bands at nearly 1357 and 1606 cm−1, assignable to the D and G band of carbon material. Another two bands at 591 and 671 cm−1 are shown in the OMC@MnO2 and OMC@MnO sample, which are attributed to the stretching vibrations of the MnO6 octahedra [31]. For the OMC@MnO sample, an extra band at 373 cm−1 is also observed, which might originate from the asymmetric stretch of bridge oxygen species (Mn–O–Mn) [32]. Figure 3c depicts the XPS spectra of the OMC, OMC@MnO2 and OMC@MnO samples. It is obvious for the OMC sample, only C1 s and O1 s were detected. For OMC@MnO2 and OMC@MnO samples, the Mn bands were clearly shown.

a XRD patterns, b Raman spectra and c XPS spectra of the OMC, OMC@MnO2 and OMC@MnO samples
The OMC, OMC@MnO2 and OMC@MnO samples were used as anode materials for lithium-ion batteries, and their electrochemical properties were evaluated via coin-type half-cells. CV measurements were carried out over a voltage range from 0.01 to 3 V (vs. Li/Li+) for three cycles to analyze the reaction mechanism of the electrode materials. The typical results are displayed in Fig. 4a–c. It is obviously seen that these three types of electrodes show different CV profiles with each other. For the OMC electrode, two obvious lithiation peaks are observed in the first cathodic scan. However, the one at around 0.5 V disappears from the second cycles, indicating the occurrence of the irreversible reactions associated with the decomposition of the electrolyte and the formation of solid-state electrolyte interface (SEI) layer. The cathodic peak near 0.01 V remains in the following lithiation processes, and the corresponding anodic peak is shown around 0.2 V during the delithiation processes, both of which are the typical characterizations for the carbon anode materials. A pair of weak redox peaks is also observed at around 1.2 V, which is probably related to the oxygen species in the carbon material. For the OMC@MnO2 electrode, the lithiation potential is located at around 0.08 V for the first cathodic scan, while it splits into two from the second cathodic sweep. The lower one at 0.01 V is contributed by the lithium insertion in OMC, while the higher one around 0.25 V by the conversion reaction of amorphous MnO2. One anodic peak is shown at 1.2 V during the delithiation scans. The CV profiles of the OMC@MnO electrode are similar to that of the OMC@MnO2 electrode. However, the splitting between the two cathodic peaks is more notable, indicating that the lithiation potential of crystalline MnO is ~ 0.1 V higher than that of the amorphous MnO2, and the anodic peak appears at 1.3 V in the delithiation process.

Typical CV profiles of a OMC, b OMC@MnO2, c OMC@MnO electrodes. Typical galvanostatic charge–discharge curves of d OMC, e OMC@MnO2, f OMC@MnO electrodes at a current density of 100 mA g−1
Figure 4d–f compares the first consecutive five galvanostatic charge–discharge curves for the OMC, OMC@MnO2 and OMC@MnO electrodes at a current density of 100 mA g−1. The initial specific discharge/charge capacities (based on the total weight of the active materials) are 917/428, 1054/613 and 829/639 mAh g−1, giving rise to the initial Coulombic efficiencies of 46.7%, 58.2% and 77%, respectively. The charge–discharge curves nearly coincide with each other from the second cycle for all three cells, indicating the good stability of these electrode materials.
The rate capability of the OMC, OMC@MnO2 and OMC@MnO electrodes was investigated by varying the galvanostatic charge–discharge current density, and the results are displayed in Fig. 5a. The average specific capacity of the OMC electrode at 100 mA g−1 is around 350 mAh g−1, close to the theoretical value of the graphite anode (375 mAh g−1). This capacity decreases quickly to approximately 140 mAh g−1 when the current density increases to 2000 mA g−1. The average capacity of the OMC@MnO2 electrode is 605 mAh g−1, nearly twofold of that of the OMC anode, indicating a significant contribution to the reversible energy storage capacity from the amorphous MnO2 nanoflakes. However, the capacity degrades greatly at higher rates, as only 23% (140 mAh g−1) is retained at 2000 mAh g−1. The OMC@MnO electrode shows a similar specific capacity (637 mAh g−1) at 100 mA g−1 as that of the OMC@MnO2 anode, but 60% (375 mAh g−1) of the capacity could be retained at 2000 mA g−1, revealing that the thermal treatment is advantageous for the electrochemical property of the OMC@MnO material. The improvement in the rate capability is probably related to the transformation from amorphous MnO2 flakes to crystalline MnO nanoparticles after the thermal treatment, which is beneficial for the structural stability and the electronic conductivity of the anode material. Figure 5b displays the cycling stability of the three anodes at 100 mA g−1 up to 100 cycles. It is seen that under the low current density, the OMC, OMC@MnO2 and OMC@MnO electrodes show an overall good cycling stability. The capacity of the OMC anode is almost remained unchanged during the cycling process; the OMC@MnO2 shows a 15% decrease of the initial value, while the OMC@MnO anode has a slight increase. When cycled at 500 mA g−1, as presented in Fig. 5c, the OMC maintains a stable capacity around 280 mAh g−1 up to 450 cycles and then slowly decreases thereafter with a declining rate of 0.13% per cycle. The capacity of the OMC@MnO2 anode shows a rapid degradation for the first 30 cycles, but it is maintained relatively stable for the following 470 cycles with an average capacity of 265 mAh g−1. The OMC@MnO anode shows a much higher capacity than that of the OMC and OMC@MnO2 anodes. As presented in Fig. 5c, there is a remarkable capacity increase from 535 to 772 mAh g−1 during the first 200 cycles, followed by a gradual decrease. The change in the capacity could be probably ascribed to the activation and subsequent degradation process of the electrode materials, associated with the structural and phase changes of the electrode upon cycling. The OMC@MnO anode exhibits an excellent cycling performance as the capacity still maintains at 434 mAh g−1 after 1000 cycles, with a 0.051% declining rate per cycle from the maximum capacity. Figure 5d displays the charge–discharge curves of the OMC@MnO electrode with normalized capacity from the 1st cycle to the 1000th cycle at 500 mA g−1 at an interval of 100 cycles. It is obvious that there are two different sloped sections of the discharge curves, indicating the existence of two lithium storage mechanisms. The section of larger slope at the higher voltage range probably corresponds to the capacitive adsorption of lithium ions, while the plateau section at lower voltage range is based on the conversion reaction of the metal oxide. As cycling proceeds, the portion of capacitive-contributed capacity increases, while the conversion-contributed capacity was reduced. This observation might be accounted for by the structural change in the MnO crystallites, which are converted to MnO nanocrystallines with greatly reduced crystal size during the repeated charge–discharge cycling. Those nanocrystallines were embedded in amorphous matrix, favorable for the surficial capacitive adsorption.

a The rate performance of the OMC, OMC@MnO2 and OMC@MnO electrodes. The cycling stability of the OMC, OMC@MnO2 and OMC@MnO electrodes at b 100 and c 500 mA g−1. d The charge–discharge curves of the OMC@MnO electrode from the first cycle to the 1000th cycle
The OMC@MnO electrode after 1000 charge/discharge cycles (ended in a full charge state) was further investigated by SEM, TEM and XRD, and the results are presented in Fig. 6. From the SEM image in Fig. 6a, it is clear that the OMC@MnO nanospheres have a significant morphological change upon prolonged cycling, but the particles still retain good integrity, which could probably account for the excellent cycle life of the OMC@MnO electrode. Figure 6b shows the TEM image of the cycled electrode material. Nanocrystallites with clear lattice fringes are observed, as indicated by the red dashed circles. Figure 6c displays the XRD patterns of the OMC@MnO electrode on copper foil before (black) and after 1000 lithiation/delithiation cycles (red). Before cycling, the peaks (except those arising from carbon conductive additives and copper foil) are readily indexed to MnO phase, which disappear after long-term cycling. However, two new diffraction peaks at 2θ = 30.4° and 31.8° are observed in the cycled electrode, as marked by black rhombuses. Though it is hard to ascertain the phase of the material via our XRD and TEM results, it is certain that some crystallinity remains in the cycled material, which is not in line with previous studies that pointed out the conversion-based metal oxide anodes that would undergo a crystalline-to-amorphous transformation during the first lithiation reaction and remain amorphous thereafter [33,34,35]. However, our XRD result provides the direct proof of a certain crystallinity of the long-term cycled electrode material. Therefore, further study is still required to reveal the cycling-dependent structural and crystallinity of the conversion-type active materials.

a SEM and b TEM images of the OMC@MnO electrode after 1000 charge/discharge cycles. The red dashed circles in b indicate the MnO nanocrystallites with legible lattice fringes. c XRD patterns of the fresh and the long-term cycled OMC@MnO electrodes on copper foil
To further study the effects of the MnO coating on the electrochemistry properties of the negative electrode, the potential polarizations ΔE (ΔE = Eanodic − Ecathodic, where Eanodic and Ecathodic are extracted from the voltage versus Li/Li+ where half capacity was attained in the charge/discharge half-cycles in this study) of the battery cells were extracted and investigated. Figure 7 shows the change of ΔE of the OMC, OMC@MnO2 and OMC@MnO electrodes as cycling proceeds (at 500 mA g−1). It is found that the OMC electrode has a small ΔE between 0.2 and 0.3 V. However, after the incorporation of a MnO2 or MnO layer, large initial ΔE about 0.9–1 V is obtained, which is a signature for the conversion-based anode materials [36,37,38]. Moreover, it rapidly increases to over 1.4 V after 400 cycles, presenting an obviously faster growing speed than that of OMC. The most interesting result is that after reaching a maximum (1.46 V around 450th cycle), the ΔE of the OMC@MnO electrode began to decrease thereafter, and the final ΔE around 1 V was obtained at 1000th cycle. This observation is quite different from our previous investigation on ZnMn2O4 electrode which displays a saturation of ΔE with cycling. This unique phenomenon about the change of ΔE for the OMC@MnO electrode is observed for the first time and might be accounted for by the changes in structure, phase or conversion/deconversion reaction pathways of the active material. However, further investigation is still needed to understand the underlying mechanisms.

The potential polarization of the OMC, OMC@MnO2 and OMC@MnO electrodes with cycle number
Conclusions
Ordered mesoporous carbon (OMC) nanospheres, MnO2-coated OMC (OMC@MnO2) nanospheres and MnO-coated OMC (OMC@MnO) nanospheres were synthesized and their morphology, nanostructures, phases and electrochemical properties as anode materials for lithium-ion batteries were investigated. The OMC@MnO2 and OMC@MnO nanospheres show remarkable improvements in specific energy capacity compared with the OMC anode. The OMC@MnO anode displays an outstanding battery performance in terms of high specific energy storage capacity, good rate capability and long cycling stability, exhibiting a cycling life up to 1000 cycles and a very low degrading rate of 0.052% per cycle. The morphology and crystallinity of the long-term cycled electrode material were investigated by SEM, TEM and XRD, which indicate a certain crystallinity of the cycled material. The potential polarization between the charge and discharge of the OMC@MnO2 and OMC@MnO electrodes is quite larger than that of OMC. The unique change in the potential polarization of the OMC@MnO electrode with long-term cycling was observed, which showed an increase and a subsequent decrease over 1000 charge/discharge cycles.
References
Li J, Li Z, Ning F et al (2018) ACS Omega 3:1675. https://doi.org/10.1021/acsomega.7b01832
Wang D, Yu Y, He H, Wang J, Zhou W, Abruña HD (2015) ACS Nano 9:1775. https://doi.org/10.1021/nn506624g
Ding C, Jiang X, Huang X et al (2018) Mater Res Bull 97:142. https://doi.org/10.1016/j.materresbull.2017.08.058
Xia T, Xu X, Wang J et al (2015) Electrochim Acta 160:114. https://doi.org/10.1016/j.electacta.2015.02.017
Huang Y, Xu Z, Mai J et al (2017) Nano Energy 41:426. https://doi.org/10.1016/j.nanoen.2017.10.001
Bai Z, Ju Z, Guo C, Qian Y, Tang B, Xiong S (2014) Nanoscale 6:3268. https://doi.org/10.1039/c3nr05676g
Gu L, Xie W, Bai S et al (2016) Appl Surf Sci 368:298. https://doi.org/10.1016/j.apsusc.2016.01.270
Jadhav HS, Thorat GM, Mun J, Seo JG (2016) J Power Sources 302:13. https://doi.org/10.1016/j.jpowsour.2015.10.044
Li L, Raji A-RO, Tour JM (2013) Adv Mater 25:6298. https://doi.org/10.1002/adma.201302915
Wei Y, Zi Z, Chen B et al (2018) J Alloys Compd 756:93. https://doi.org/10.1016/j.jallcom.2018.04.331
Chen J, Wang Y, He X et al (2014) Electrochim Acta 142:152. https://doi.org/10.1016/j.electacta.2014.07.089
Jian G, Xu Y, Lai L-C, Wang C, Zachariah MR (2014) J Mater Chem A 2:4627
Shi S, Deng S, Zhang M, Zhao M, Yang G (2017) Electrochim Acta 224:285. https://doi.org/10.1016/j.electacta.2016.12.080
Gu X, Yue J, Li L, Xue H, Yang J, Zhao X (2015) Electrochim Acta 184:250. https://doi.org/10.1016/j.electacta.2015.10.037
Su H, Xu Y-F, Feng S-C et al (2015) ACS Appl Mater Interfaces 7:8488. https://doi.org/10.1021/am509198k
Wang J, Liu Y, Wang S, Guo X, Liu Y (2014) J Mater Chem A 2:1224. https://doi.org/10.1039/C3TA14135G
Zhang R, Liu J, Guo H, Tong X (2015) Mater Lett 139:55. https://doi.org/10.1016/j.matlet.2014.10.039
Kim J, Hong S-A, Yoo J (2015) Chem Eng J 266:179. https://doi.org/10.1016/j.cej.2014.12.084
Huang XH, Guo RQ, Wu JB, Zhang P (2014) Mater Lett 122:82. https://doi.org/10.1016/j.matlet.2014.02.012
Poizot P, Laruelle S, Grugeon S, Dupont L, Tarascon J (2000) Nature 407:496
Shao J, Zhou H, Zhu M, Feng J, Yuan A (2018) J Alloys Compd 768:1049. https://doi.org/10.1016/j.jallcom.2018.07.290
Sheng L, Liang S, Wei T et al (2018) Energy Storage Mater 12:94. https://doi.org/10.1016/j.ensm.2017.11.014
Zhao Y, Huang G, Li Y et al (2018) J Mater Chem A 6:7227. https://doi.org/10.1039/C8TA00940F
Fan Z, Liang J, Yu W et al (2015) Nano Energy 16:152. https://doi.org/10.1016/j.nanoen.2015.06.009
Xiao S, Pan D, Wang L et al (2016) Nanoscale 8:19343. https://doi.org/10.1039/C6NR07802H
Dou Y, Xu J, Ruan B et al (2016) Adv Energy Mater 6:1501835. https://doi.org/10.1002/aenm.201501835
Gu D, Li W, Wang F et al (2015) Angew Chem Int Ed 54:7060. https://doi.org/10.1002/anie.201501475
Zhou G, Wang D-W, Li F et al (2010) Chem Mater 22:5306. https://doi.org/10.1021/cm101532x
Xiao Y, Wang X, Wang W, Zhao D, Cao M (2014) ACS Appl Mater Interfaces 6:2051. https://doi.org/10.1021/am405142p
Li M, Xue J (2012) J Colloid Interface Sci 377:169. https://doi.org/10.1016/j.jcis.2012.03.085
Gao T, Fjellvåg H, Norby P (2009) Anal Chim Acta 648:235. https://doi.org/10.1016/j.aca.2009.06.059
Ramesh K, Chen L, Chen F, Liu Y, Wang Z, Han Y-F (2008) Catal Today 131:477. https://doi.org/10.1016/j.cattod.2007.10.061
Kim S-W, Lee H-W, Muralidharan P et al (2011) Nano Res 4:505. https://doi.org/10.1007/s12274-011-0106-0
Wang GX, Chen Y, Konstantinov K, Lindsay M, Liu HK, Dou SX (2002) J Power Sources 109:142. https://doi.org/10.1016/S0378-7753(02)00052-6
Deng Y, Li Z, Shi Z, Xu H, Peng F, Chen G (2012) RSC Adv 2:4645. https://doi.org/10.1039/C2RA20062G
Taberna P-L, Mitra S, Poizot P, Simon P, Tarascon J-M (2006) Nat Mater 5:567
Malini R, Uma U, Sheela T, Ganesan M, Renganathan NG (2009) Ionics 15:301. https://doi.org/10.1007/s11581-008-0236-x
Mai YJ, Shi SJ, Zhang D, Lu Y, Gu CD, Tu JP (2012) J Power Sources 204:155. https://doi.org/10.1016/j.jpowsour.2011.12.038
Acknowledgements
The author gratefully acknowledges Professor Paul V. Braun of Department of Materials Science and Engineering, University of Illinois at Urbana–Champaign (UIUC), for his useful advice and the use of his laboratory for this research wok. The author also acknowledges Li Zhao for his assistance in the acquisition of XRD patterns. The author also acknowledges Chinese Scholarship Council during her visit to UIUC, and partial support from National Natural Science Foundation of China (21503036). The research was carried out in the Frederick Seitz Materials Research Laboratory Central Research Facilities, University of Illinois at Urbana–Champaign.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Feng, T., Wang, J., Yang, J. et al. Investigation of ordered mesoporous carbon@MnO core–shell nanospheres as anode material for lithium-ion batteries. J Mater Sci 54, 6461–6470 (2019). https://doi.org/10.1007/s10853-018-03307-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-018-03307-5