
Abstract
A high-performance aromatic co-polyimide (co-PI) fiber was prepared by thermal treating the polyamic acid (PAA) precursor fiber produced through a wet spinning technique. A gradient thermal treatment protocol, which was selected based on the thermogravimetric analysis of PAA fibers, was employed for achieving the best structure and fiber performance. The structural and morphological variations of fibers at different thermal treatment stages were verified by Fourier transform infrared spectroscopy, sonic orientation detection, wide-angle X-ray diffraction and scanning electronic morphology. Thermal treatment cycloimidizes the PAA precursor into PI fibers with the concomitant development of a laminar orientation structure expanded from outer to inner layers of the fiber. The ordered chain repeat length is calculated to be 1.62 nm in the meridian direction. Mechanical measurements indicate that the synthesized co-PI fiber achieves optimum fracture strength and initial modulus up to 3.56 and 101 GPa, respectively. Thermal characterization results indicate a 5% weight loss temperature under nitrogen up to 591 °C and a glass transition temperature at 340 °C.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyimide (PI) constitutes a class of high-performance aromatic heterocyclic materials with versatile applications from aerospace to biologicals due to its outstanding thermal stability, chemical and radiation resistance, as well as the extraordinary mechanical and dielectric properties [1,2,3,4,5,6,7]. As one of the most important material forms, PI fiber has gained widespread interests in both fundamental and industrial research fields since 1980s when Soviet researcher Koton et al. [8,9,10,11,12,13,14,15,16] made a series of different PI fibers. PI fibers based on dianhydride of diphenyloxidetetracarboxylic acid and pyromellitic acid and various diamines containing phenylene oxide, diphenylmethane, p-phenylene, fluorene and diazo groups were prepared. Also, PI fibers containing heterocyclic units (e.g., pyridine, oxadiazole, benzimidazole) in the diamine and acid dianhydride fragments were prepared. Accordingly, extensive researches associated with the structure and properties of PI fibers were performed [9, 11, 14]. And great achievement was attained by Stephen Z. D. Cheng during 1990s based on his detailed studies with the wide-angle X-ray diffraction (WAXD), thermogravimetric analysis (TGA), dynamic mechanical analysis (DMA), scanning electronic morphology (SEM) and tensile stress–strain investigations on a series of homo-polyimide (homo-PI) and co-polyimide (co-PI) fibers prepared via a one-step technique [17,18,19,20,21,22,23,24,25,26,27] .
Currently, there are two principal techniques for preparing PI fibers, namely one-step method and two-step method. Generally, due to the incomplete cyclodehydration of PAA fibers in the two-step technique, the one-step approach is more favorable for producing high-performance PI fibers since the possibility for forming defects, which is always the major problem in the two-step technique, could be completely eliminated [5,6,7, 28,29,30]. As an example, Cheng et al. [2] have successfully prepared high-performance 3,3′,4,4′-biphenyltetracarboxylic dianhydride/2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl (BPDA/PFMB) PI fibers via one-step method. The resultant PI fibers with 10 times draw ratio display a tensile strength of about 3.2 GPa and an initial modulus higher than 130 GPa. The BPDA/PFMB PI fibers also display an excellent thermal stability because it shows a 5% weight loss under dry nitrogen and air atmosphere at 600 °C. However, only limited number of PI fibers can be prepared by one-step method due to the difficulty in finding powerful solvents for dissolving PIs with high molecular weight. Most of these fibers are derived from the 3,3′,4,4′-biphenyltetracarboxylic dianhydride due to its higher solubility in phenolic solvent [31, 32], such as phenol, p-chlorophenol, m-cresol, p-cresol and 2,4-dichlorophenol. Consequently, the two-step technique, through which PI fibers are produced by thermal treatment of the polyamic acid (PAA) precursor fibers spun from PAA solution, becomes important and turns into the focus of research interests in recent years [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54]. The thermal treatment protocol plays a key role in preparing high-performance PI fibers since porous or skin–core structure would be readily formed if inappropriate thermal process is applied [37, 45, 55,56,57].
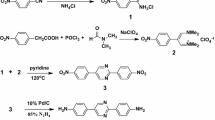
In this paper, we report our work on preparation of a novel high-performance co-PI fiber from its precursor PAA solution via the two-step technique by employing an experimentally determined gradient thermal treatment protocol. The PAA solution is prepared by condensation polymerization of 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA), 2-(4-aminophenyl)-5-aminobenzimidazole (BIA) and p-phenylenediamine (p-PDA) in dimethylacetamide (DMAc) solvent. The reaction process and chemical structures are shown in Scheme 1. Thermal treatment converts the PAA fiber into its final PI form with the concomitant establishment and elaboration in orientation structure and morphology, endowing the PI fibers with considerably improved mechanical properties. The gradual variations of orientation structure were investigated by measuring the WAXD patterns and sonic orientation responses of the fibers produced at different thermal treatment stages. The surface and cross-sectional morphologies of the fibers were traced during the whole thermal stage by SEM observation. Thermal properties of the final PI fibers were characterized by TGA and DMA.

Synthesis of the BPDA/BIA/p-PDA co-PAA and co-PI
Experimental
Materials and fiber spinning
PAA solution was prepared by condensation polymerization of 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA), p-phenylenediamine (p-PDA) and 2-(4-aminophenyl)-5-aminobenzimidazole (BIA) in dimethylacetamide (DMAc) solvent with a 15% (w/w) solid concentration, and the molar ratio of BPDA, BIA and p-PDA is 4:1:3. The intrinsic viscosity of PAA solution at 35 °C was 3.2 dL/g. Prior to use, the synthesized PAA precursor solution was degassed under vacuum at 50 °C to remove the existing air. PAA fibers were then prepared through the wet spinning process by extruding the degassed PAA solutions through a spinneret (100 holes with 70-μm hole diameter) under nitrogen pressure of 0.2 MPa into a coagulation bath filled with deionized water. The extruded fibers were immediately drawn under constant speed (0.5 m/min) and finally collected on a roller for further treatment.
Thermal treatment
The cyclodehydration of PAA fibers to PI fibers was carried out in a horizontal electric furnace with a 1.4-m-long ceramic tubular chamber (22 mm in diameter). A bundle of PAA fibers was drawn out from a feeding roller, admitted through the furnace and wounded on a reel placed behind the outlet of the furnace. The feeding roller and the fiber collecting roller were set at the same speed, so there was no tension during thermal treatment process. The thermal treatment time of the fibers in the furnace was calculated to be 2 min. The fibers were treated under four gradient thermal stages, i.e., 80 °C for 2 min, 240 °C for 2 min, 300 °C for 2 min and 370 °C for 2 min. The temperature of the last thermal stage was set above the Tg of the PI and varied from 340 to 380 °C during the experiment. Finally, 370 °C was selected for the thermal cycle since optimum mechanical properties were achieved on the PI fibers at this temperature.
Characterization
Mechanical properties of the fibers were tested on a filament tensile instrument manufactured by Tai Cang Hong Da Fang Yuan Electric Co., Ltd, in Chang Zhou, China. The gauge length and tensile speed were 20 mm and 10 mm/min, respectively. The diameter of the filament was determined from the microscopic observation (Motic group BA200 microscope). For each sample, at least 10 filaments were tested and the average values were reported as the mechanical property.
Densities of PAA and PI fibers with different thermal treatments were measured with a GH-300S density meter produced by Matsuhaku Company in Taiwan. Firstly, fibers were ultrasonic cleaned in acetone for 30 min to remove the surface contaminants. Then, the ultrasonic cleaned fibers were vacuumed and dried at 60 °C for 30 min to be weighed as m 1. Secondly, the fibers were ultrasonic cleaned in acetone for 30 min again to exclude the air bubbles between fibers. Then, the ultrasonic cleaned fibers were washed by ethanol and placed in density meter loaded ethanol to be weighed as m 2. Finally, the density of fibers can be calculated using the following equation:
where ρ f represents the density of fibers and the unit is g/m3, and ρ l represents the density of liquid in density meter and the unit is g/m3.
Fourier transform infrared (FTIR) spectra of the fibers treated at different thermal stages were collected on a Nicolet Nexus 670 FTIR spectrometer in the wave number range of 4000–400 cm−1.
Two-dimensional wide-angle X-ray diffraction (2D WAXD) patterns were recorded on a Bruker D8 Discover diffractometer equipped with a general area detector diffraction system (GADDS) as a 2D detector, in a transmission mode at room temperature. X-ray generator uses Cu Kα radiation (0.1542 nm). The peak positions were calibrated using the diffraction of silicon powder (2θ = 28.47°). A bundle of closely stacked parallel fibers were fixed on the specimen holder by double-sided adhesive tape, and the point-focused X-ray beam was aligned perpendicular to fiber axis.
The sonic velocity in the fibers was measured on a SCY-I-type sonic orientation detector manufactured by the Material Academe of Donghua University, China. The length of the fiber samples was about 70–80 cm, and the measurement was taken on different points along the fibers by moving the receiver to change the testing distance. For each fiber sample, 8–10 data points were collected and linearly fitted to give the sonic velocity.
SEM images were recorded on a SEM-4500 microscope (JEOL Ltd, Tokyo, Japan) and a quanta scanning electron microscope (FEI Quanta 200, Czech Republic). The surface morphology was observed by mounting the fibers in short length on a specimen holder using conductive adhesive. For the cross-sectional observation, the fibers were embedded in epoxy resin followed by brutally broken in liquid nitrogen to give the freeze-fractured cross sections or cut with a sharp razor blade to yield the flat cross sections. All the samples prepared for both surface and cross-sectional morphology observations were coated with a ca. 5-nm gold layer prior to measurement.
TGA measurements of both PAA and PI fibers were taken on a TGA Q50 system (TA Company, USA) at a heating rate of 10 °C/min. Glass transition temperature (Tg) of PI fiber was determined with the DMA system of Q800 (TA instrument) at a heating rate of 5 °C/min.
Results and discussion
Determination of thermal treatment protocol
Figure 1 exhibits the TGA and DTG thermal behavior of the as-spun PAA fibers. With the increase in the temperature, the TGA curve exhibits four distinct weight loss stages in the range of 25–110 °C (stage I), 110–240 °C (stage II), 240–300 °C (stage III) and 300–380 °C (stage IV). The weight loss in stage I was determined to be ca. 4.89 wt%, which is suggested to be the removal of the free water molecules diffused into the PAA fibers during the coagulation process. Further thermal treatment evaporates the remaining solvents (mainly DMAc) in the fibers and simultaneously converts the PAA fiber into its imide form through cycloimidization. Figure 1 demonstrates that the total weight loss in stage II, stage III and stage IV is 10.1 wt%, which corresponds to the removal of the existing solvents and the elimination of water molecules during the polymer cyclization process. Theoretical calculation suggests that the weight loss of PAA caused by complete cyclodehydration is 9.22 wt%. Thus, the residual solvents in the PAA fibers are estimated to be 0.88 wt% by subtracting the cyclodehydration weight loss of PAA (9.22 wt%) from the total weight loss (10.1 wt%) in stage II, stage III and stage IV. Experimental results indicate that fibers treated between 150 and 200 °C become considerably brittle and easily fractured into short pieces without any mechanical usefulness, which probably results from the degradation of PAA chains. However, at enhanced temperature over 200 °C, the fibers become flexible again and grow stronger with increasing temperature, mainly owing to the occurrence of cycloimidization. From Fig. 1, we can see that the weight loss of 7.57 wt% in stage II is occurring in the range of 110–240 °C. Because the residual solvents in PAA fibers are calculated only to be 0.88 wt%, the 7.57 wt% weight loss suggests the achievement of a considerably high degree of imidization in the fibers when treated to 240 °C, consistent with the 90.4% degree of imidization obtained from FTIR analysis (shown later in Fig. 4). Subsequent thermal treatment at stage III and stage IV only brought a 2.53% total weight loss, which denoted a slow and difficult imidization process. Nevertheless, degree of imidization of PI fibers was enhanced to 92.5 and 100% (calculated from FTIR spectra in Fig. 4) after thermal treatment at stage III and stage IV, respectively. Thus, thermal treatment at stage III and stage IV is essential and tremendously significant for achieving high-performance PI fibers since it is believed that what occurs at these two stages is mainly the perfection of the imidization and the regularity of the PI structure. Based on the above analysis, a four-step gradient thermal treatment process was finally determined for the cyclization of the PAA fibers to produce the high-performance PI fibers, i.e., 80 (step I), 240 (step II), 300 (step III) and 370 °C (step IV). Step 1 was referred to be the pretreatment stage, and 80 °C was selected for removing the adsorbed free water molecules to avoid the possible formation of defects in the fibers during the subsequent high-temperature dehydration process. The temperature of 240 °C was selected as the second step since predominant imidization would take place at this temperature and the degradation of PAA chains between 150 and 200 °C could be effectively circumvented. For step III and step IV, 300 and 370 °C were employed respectively with the purpose of realizing perfect imidization and structure regularity. Work on the successful preparation of high-performance PI fibers with this gradient thermal annealing process is shown later.

TGA and DTG curves of PAA fiber measured under nitrogen atmosphere
Thermal behaviors of the PI fibers
Figure 2 exhibits the TGA curves for the final PI fibers measured under air and nitrogen atmosphere. The data demonstrate the excellent thermal stability of our prepared PI fibers with a 5% weight loss up to 579 and 591 °C under air and dry nitrogen, respectively, which is highly superior to that of BPDA–ODA [58] and PMDA–ODA–PABZ PI fibers [28] and comparable to that of the BPDA–PFMB PI fibers (600 °C) [2].

TGA curves of the PI fibers measured under air and dry nitrogen atmosphere
The thermal dynamic behaviors of the PI fibers were presented by its DMA characteristics in Fig. 3. As can be observed, the fiber exhibits clear thermal relaxation behavior during the temperature scanning process, with the appearance of two distinct peaks on the tan–delta curve. The peak located at 340 °C corresponds to the Tg of the PI fibers and is generally named as α-relaxation [23, 24], which is suggested to originate from the segment motion of the polymer chains. The peak centered at 207 °C with a broad range from 100 to 250 °C is referred to be β-relaxation [59, 60] and interpreted as the non-cooperative motion of the uncrystallized diamines in the co-PI fibers based on Li’s report [23].

DMA curves of PI fibers with a heating rate at 5 °C/min and frequency at 1 Hz
Mechanical properties of the fibers
By utilizing the gradient thermal annealing protocol, PI fibers with excellent mechanical properties were prepared in our present work. As a summary, Table 1 lists the variation of the various properties of the PI fibers during the thermal treatment process, including diameter, density, fracture strength, initial modulus and percentage elongation at break.
With the proceeding of the thermal annealing process, the diameter of the sample fiber decreased gradually from 16.2 μm for the as-spun PAA fiber to 12.5 μm after being treated to 370 °C. This fiber thinning down phenomenon observed during thermal curing is under expectation since solvent evaporation and cyclodehydration are known to occur. And it is suggested that the diameter changes occurring before 240 °C are mainly due to solvent removal, while further diameter decrease observed after that is primary caused by further cycloimidization of PAA and the subsequent dense packing effect of polymer chains at elevated temperature [57, 61]. The fiber diameter holds constant at 12.5 μm during 350–370 °C. However, it exhibits a non-ignorable augmentation to 13.7 μm after raising the temperature to 380 °C. This is unexpected and is suggested to arise from the disorientation of polymer chains at high temperatures to increase the entropy elasticity.
The density of fibers increases from 1.288 to 1.408 g/cm3 gradually with temperature increasing from 80 to 370 °C and then decreases dramatically to 1.32 g/cm3 when temperature increasing to 380 °C. The increase in density of fibers arises from solvent removal and gradual cycloimidization of PAA, which can promote the formation of dense packing and ordered polymer chain. The decrease in density of PI fibers when the temperature is above 370 °C is mainly caused by the disorientation of polymer chains. The trend of change of density is in accordance with the fracture strength during the gradient thermal treatment process.
Fracture strength increases dramatically with temperature and reaches a maximum value of 3.56 GPa at 370 °C. The data shown in Table 1 indicate that pretreatment at 80 °C has limited effects on the fibers’ overall mechanical performances. The steepest and vast enhancement in fracture strength was observed on the fibers treated at 240–300 °C, corresponding to the formation of rigid heterocyclic structures due to thermal imidization of PAA and the formation of highly oriented polymer chains along the fiber axis, which can be observed in 2D WAXD patterns (shown in Fig. 5). Further thermal treatment at Tg (340 °C) or higher temperature (350, 360 and 370 °C) results in a further significant enhancement in fracture strength, and the best performance was realized at 370 °C. The promotion of fracture strength at this stage is suggested to be the perfection of cycloimidization [57, 61], as determined by infrared analysis, together with the perfection of highly oriented laminar structure [54] of polymer chains along the fiber axis (revealed by SEM in Fig. 7) due to gradient elevated temperatures treatment. The variation of the fibers’ initial modulus follows a similar trend during the thermal curing process but with an apparent decrease from 133 GPa to around 101 GPa when treated at the temperature above Tg.
For the elongation at break for the as-spun PAA fibers, pretreatment at 80 °C reduces it from 9.0 to 3.6%. And, it exhibits only a slight variation in the range of 2.1–3.6% during the followed thermal curing process. The relatively low percentage elongation suggests the rigid characteristics of the obtained fiber.
The above-mentioned results suggest that thermal treatment protocol has a considerably significant influence on the fibers’ performance and the thermal annealing above Tg is essential for achieving high-performance PI fibers. In the present study, the optimum PI fibers were prepared with fracture strength of 3.56 GPa, modulus of 101 GPa and elongation at break of 3.5% under a gradient thermal protocol of 80–240–300–370 °C. Thermal treatment at or above 380 °C in air should be strictly avoided since disorientation of polymer chains in polyimide fibers would occur and consequently results in a serious compromise of the fibers’ mechanical properties, as shown in the last row in Table 1.
The mechanical properties of PI fibers prepared under our present experimental conditions are mostly related to the chemical structure, orientation and morphology. After thermal cycloimidization at 240 °C, a rigid-rod PI structure is basically formed and multiple hydrogen bonds are also formed intermolecular due to benzimidazole units in BIA [42, 43], which are the foundation of preparation of high-performance co-polyimide fibers. Besides, a highly ordered orientation structure of polymer chains along the fiber axis is formed at the same time according to WAXD patterns (shown in Fig. 5) and the ordered laminar fibrils can be observed clearly in SEM images (shown in Fig. 7), which account for the sudden and vast enhancements in fracture strength and initial modulus. When the temperature of thermal treatment is further increased from 240 to 370 °C, the perfection in rigid-rod PI chemical structure, hydrogen-bonding interaction, dense laminar fibrils and ordered orientation structure of polymer chains is further enhanced, which result in a synergetic effect on final mechanical properties of co-polyimide fibers. After thermal treatment temperature is increased, the degree of imidization is increased based on FTIR in Fig. 4, and the orientation structure of polymer chains along the fiber axis is more ordered based on the sharper and shorter streaks along the meridian direction in WAXD patterns in Fig. 5, and the laminar fibrils are more dense and homogeneous distributed in the whole cross section based on SEM images in Fig. 7. Thus, the mechanical properties of co-polyimide fibers under the gradient thermal protocol adopted in this paper are mainly affected by chemical structure of polyimide, hydrogen-bonding interaction intermolecular, orientation of polymer chain along the fiber axis and the inner morphology of laminar fibrils.

FTIR spectra of fibers at different thermal treatment processes: (a) PAA, (b) PI-240, (c) PI-300, (d) PI-380

2D WAXD pattern of fibers with different heat treatment processes: a PAA, b PAA-80, c PI-240, d PI-300, e PI-340, f PI-350, g PI-360, h PI-370, i PI-380
Study of the cycloimidization process
Figure 4 exhibits the FTIR spectra of the fibers treated under different thermal protocols. Significant changes were observed when the PAA fibers were treated to 240 °C. However, after that, the FTIR spectra were almost identical, which is similar to that reported by Saeed and Xu [61, 62].
For the spectrum of the as-spun PAA fibers shown in Fig. 4a, the peak located at 1650 cm−1 is pertaining to the stretching vibration of C=O in the carboxylic acid and amide groups. The appearance of this peak suggests the amic acid characteristics of the fibers.
The four distinct absorbances at 1773, 1718, 1354 and 737 cm−1 in Fig. 4b suggest that PAA was cycloimidized into PI after thermal treatment at 240 °C. The peaks at 1718 and 1773 cm−1 are corresponding to the C=O symmetric and asymmetric stretching vibration of the imide ring, respectively, the peak at 1354 cm−1 is related to C–N stretching vibration of the imide ring, while the peak at 737 cm−1 is related to the imide ring bending [44].
Further thermal treatment results in increase in the degree of imidization. The increase can be reflected by enhanced absorbance at 1718 and 1354 cm−1 and the weakened absorbance at 1650 cm−1 on the IR curves shown in Fig. 4c, d.
To quantitatively evaluate the variation of imidization during the thermal treatment process, the degree of imidization for the fibers treated at different temperatures was calculated by comparing the relative absorbance intensity of the peaks centered at 1354 cm−1 (νC–N of imide ring) and 1513 cm−1. The absorbance at 1513 cm−1 is ascribed to the C–C stretching of p-substituted benzene and selected as the internal standard since it is supposed to be constant during the whole thermal procedure [61, 62]. The fibers treated at 380 °C are assumed to be cyclized completely and used as the external standard with 100% imidization. Thus, the degree of imidization of the fibers heated at selected temperature could be calculated using the following equation:
where D1354 and D1513 represent the IR absorbance intensity of the peaks at 1354 and 1534 cm−1, respectively; T and 380 represent the thermal treatment temperature of the sample fibers and the external standard fibers.
Calculation results indicate that about 90.4% imidization has already been accomplished in the fibers after heated at 240 °C. However, it becomes rather slow after that and subsequent thermal treatment at 300 °C only brings about limited increase in the degree of imidization from 90.4 to 92.5%. The limited increase in the degree of imidization is considered to be caused by severely restricted chain mobility after cyclization. Hence, to realize complete imidization for producing high-performance PI fibers, an elevated temperature over Tg is required in the thermal procedure.
As a standard method, sonic velocity detection was usually employed for observing changes in structure or arrangement and packing state of molecules, because sound wave is transmitted by stretching of chemical bonds in the backbone along the direction of polymer chain and by the way of non-bonded intermolecular vibration in the direction perpendicular to the polymer chain.
Table 2 lists our sonic velocity measurements on the fibers treated under different thermal cycles. The data indicate that the sonic velocity increases gradually with the thermal treatment temperature, which is believed to be caused by the structural variation from the relatively flexible PAA to the more rigid heterocyclic PI units. The steepest increase in sonic velocity in PI fibers was observed after thermal treatment at 240 °C (as shown in Table 2), implying the realization of a considerably high degree of imidization and orientation of polymer chains at this point, consistent with the results of mechanical properties test (Table 1) and infrared analysis (Fig. 4), whereas further thermal treatment to 300 °C only results in a slight enhancement in sonic velocity, demonstrating a slight structural variation from 240 to 300 °C. Sharp promotion in sonic velocity occurs again at the elevated temperature (370 °C), which is suggested to be caused by the perfection of imidization and high orientation of polymer molecules along the fiber axis at a temperature above Tg. The sonic velocity variation suggests the structural variation of the fibers during the thermal annealing process.
Study of the orientation and crystallization structures
Figure 5 exhibits the 2D WAXD patterns for the fibers treated at different thermal stages.
For the as-spun PAA fibers and those treated at 80 °C, Fig. 5a, b exhibits nearly identical patterns without any characteristic diffraction, implying an amorphous structure in the fibers. For the fibers treated with temperatures higher than 240 °C, seen from Fig. 5c–i, two clear diffraction streaks appear in the meridian direction, indicating the formation of the ordered structure along the fiber axis. The obscure amorphous halos along the equator direction demonstrate the poor ordered lateral packing structures of PI chains. Moreover, there is no evidence of diffraction in the quadrants, revealing that the PI fibers do not exhibit well-defined 3D crystalline structures [34, 44].
For details, the WAXD patterns along the equator and meridian directions of the fibers treated at temperatures higher than 240 °C are presented in Fig. 6a, b, respectively. The broad peaks in the range of 15–25° can be observed in the equator, representing a typical amorphous behavior of polymer chains in the transverse direction of the fibers. The intensities of the peaks increase gradually with increasing temperature, implying the improved lateral packing order of the polymer chain.

Set of WAXD fiber patterns scanning along the equatorial (a) and meridian (b) direction
In the meridian direction as shown in Fig. 6b, two peaks at about 10.9° and 15.8° with d-spacing values of 0.81 and 0.56 nm (by Bragg equation) can be observed. According to our previous work [44], the minimum energy conformation for BPDA/PDA units is supposed to have a repeat of 1.587 nm, which is close to the twice and three times of the calculated data of 0.81 nm and 0.56 nm. Therefore, the peaks at 10.9° and 15.8° are assigned to the (002) and (003) layers, respectively. The ordered chain repeat length in the meridian direction is twice as large as the d-spacing of (002) layer and is calculated to be 1.62 nm. The molecular orientation of the ordered structure along the meridian direction can be determined through an azimuthal integration of the intensity belonging to the (002) plane since it is isolated along the meridian direction. According to the results in Table 3, the degree of molecular orientation increases with increasing temperature and reaches the maximum at 370 °C. This improvement is ascribed to the formation of ordered structures during imidization process, making a contribution to the enhancement of the mechanical properties of the PI fibers. However, further increases in the temperature lead to the slight decrease in the orientation degree, which is mainly due to the decomposition of the PI backbones at high temperatures.
The freeze-fractured cross-sectional morphologies for the fibers treated under different thermal cycles are presented in Fig. 7. The cross-sectional morphology varies drastically during the thermal treatment protocol. The as-spun PAA fibers exhibit rather flat and smooth cross sections (in Fig. 7a), suggesting a brittle fracture behavior, accounting for the fibers’ poor mechanical properties at this point. Figure 7b exhibits that the cross section becomes rough after thermal treatment at 80 °C. And this phenomenon becomes considerably obvious at 240 °C, with the pulling out of laminar fibrils on the cross section (Fig. 7c), indicative of a ductile rupture. This is suggested to originate from the occurrence of thermal imidization and the subsequent formation of a hard-wall-soft-core fiber structure [54], which is consistent with the drastically increased facture strength and initial modulus. Careful observation suggests that the laminar fibrils in the outer wall are more than those in the core section. The diagrammatic sketch of cross section of fibers is shown in Fig. 7g.

Fractured cross-sectional morphological photographs of fibers at different stages of the thermal treatment process: a PAA, b PAA-80, c PI-240, d PI-300, e PI-370, f PI-370 and g diagrammatic sketch of cross section of fibers
Further thermal treatment results in a further considerable promotion in fracture strength and initial modulus with the concomitant observation of highly fibrillated structures on the fractured surfaces of the fibers treated at elevated temperature, as shown in Fig. 7d–f. In particular, for the fibers treated at 370 °C, the fibrils exhibit a homogeneous distribution in the whole cross-sectional area and the boundary between outer wall and inner core completely disappeared, which is a result of the combination of high degree of imidization and orientation of polymer chains in PI fibers. Consequently, an optimum mechanical performance was realized based on this uniform and highly fibrillated structure.
During the process of thermal treatment, the feeding roller and the fiber collecting roller on both end of the furnace are set in the same speed, so drawing ratio is 1 and stretch is not applied in thermal treatment process. Thus, stretch is not the factor accounting for the high orientation of polymer chain along the fiber axis (shown in Fig. 5). Then, rearrangement of polymer chains along the fiber axis and the consequent ordered fibrils formed along the fiber axis is speculated as the primary cause when solvent is removed during thermal treatment. During the process of thermal treatment, polymer chains have a better opportunity to arrange themselves into defect-free positions to form ordered molecular alignment due to the removal of micromolecules [33], so gradient thermal treatment would improve the mechanical properties of PI fibers [37, 48]. When solvent inside the fibers is removed, there is more free volume for the segments of polymer chains to rearrange. Although no stretch is applied in thermal treatment process, fibers are in a straight state with drawing ratio of 1. The straight state of fibers in thermal treatment would promote the ordered arrangement of segments of polymer chains along the fiber axis, and large-scale ordered fibrils are formed consequently from out wall to core of the fibers (shown in Fig. 7). After the solvent is removed during thermal treatment, diameter of fiber is smaller and the structure of fibers is denser, so the arrangement of fibrils is denser and distributed homogeneously, which accounts for the high performance of the prepared co-polyimide fibers. When the temperature of thermal treatment is increasing from 240 to 370 °C, the fibrils formed along the fiber axis are more ordered across the whole transverse cross section of fibers (shown in Fig. 7), which explained the enhancement of mechanical properties of co-polyimide fibers.
Study of the surface and cutting cross-sectional morphology
The surface characteristic of the sample fiber A, B, C, D and H (in Table 1) obtained under different thermal protocols is shown in Fig. 8. As shown, all the fibers exhibit uniform and regular surface morphologies without the appearance of defects and voids on the fiber surfaces excluding the observation of continuum notches along the longitudinal axis, which were caused innately by spinneret during the fiber spinning process. Fibers subject to thermal treatment exhibit smoother surface morphologies as compared to the as-spun PAA fibers (Fig. 8a). Figure 8f shows a bundle of PI fibers obtained at 370 °C, the image of which indicates that our prepared fibers are considerably uniform and has a diameter of about 12.5 μm.

Surface morphological photographs of fibers in different thermal treatment processes: a PAA, b PAA-80, c PI-240, d PI-300, e PI-370, f PI-370
To find the possible defects formed in the inner area, the fibers were embedded in epoxy resin and cut with a razor blade for cross-sectional observation, the results of which are shown in Fig. 9. As can be observed, voids and other defects are completely absent from the inner area of the fibers. In particular, instead of the formation of a porous structure, the fibers prepared under current procedure exhibit rather condensed cross sections, indicating the nice validity of the selected gradient thermal treatment protocol used in the present study.

Cutting cross-sectional morphological photographs of fibers at different thermal treatment processes: a PAA-80, b PI-240, c PI-300, d PI-340, e PI-350, f PI-360
Conclusions
A new high-performance aromatic co-PI fiber has been prepared via the two-step technique by employing a well-selected gradient thermal treatment protocol. The optimum fibers were prepared with fracture strength of 3.56 GPa and initial modulus of 101 GPa after thermal treatment at 370 °C, which concomitantly exhibit rather condensed and defect-free inner structures with uniform diameters of about 12.5 μm. Orientation structure formed in the fibers was confirmed by WAXD, and the repeat unit length along the meridian direction is estimated to be 1.62 nm. Thermal analysis suggests excellent thermal stability for the final PI fibers with 5% weight loss temperatures up to 591 °C under nitrogen and 579 °C in air and a glass transition temperature at 340 °C, which meet the requirements for a large amount of practical applications.
References
Ambroski LE (1963) H-Film—a new high temperature dielectric. Ind Eng Chem Prod Res Dev 2:189–194
Cheng SZD, Wu ZQ, Eashoo M, Hsu SLC, Harris FW (1991) A high-performance aromatic polyimide fibre: 1. Structure, properties and mechanical-history dependence. Polymer 32:1803–1810
Eashoo M, Buckley LJ, Clair AKS (1997) Fibers from a low dielectric constant fluorinated polyimide: solution spinning and morphology control. J Polym Sci Part B Polym Phys 35:173–185
Irwin RS, Sweeny W (1967) Polyimide fibers. J Polym Sci Part C Polym Symp 19:41–48
Mark HF (1977) Polymers for extreme service conditions. Macromolecules 10:881–888
Sasaki I, Itatani H, Kashima M, Yoshimoto H, Yamamoto S, Sasaki Y (1981) Aromatic polyimide resin composition. US Patent 4247443
Song YJ, Meng SH, Wang FD, Sun CX, Tan ZC (2002) Thermochemical study on the properties of polyimide BPADA–m-PDA. Thermochim Acta 389:19–24
D’Yakonova NV, Mikhailova NV, Sklizkova VP, Baranovskaya IA, Baklagina YG, Kudryavtsev VV, Sidorovich AV, Eskin VY, Koton MM (1986) Initial stages of structurization in dilute solutions of polyamic acids and polyimides. Polym Sci USSR 28:2646–2653
Korzhavin LN, Prokopchuk NR, Baklagina YG, Florinskii FS, Yefanova NV, Dubnova AM, Frenkel SY, Koton MM (1976) Correlation of chain configurations, structure and mechanical properties of fibres of polypyromellitimide series. Polym Sci USSR 18:807–814
Koton MM (1977) Intramolecular cyclization of linear polymer molecules. Review. Polym Sci USSR 19:1627–1639
Koton MM (1979) The synthesis, structure and properties of aromatic polyimides (PI). A review. Polym Sci USSR 21:2756–2767
Koton MM, Kiseleva TM, Zhukova TI, Nikolayeva SN, Laius LA, Sazanov YN (1981) Polyimides containing various heterocyclic main-chain units. Polym Sci USSR 23:1909–1915
Koton MM, Sazanov YN (1975) Thermal degradation of aromatic polyimides based on diphenyloxidetetracarboxylic acid. Polym Sci USSR 17:1688–1697
Prokopchuk NR, Baklagina YG, Korzhavin LN, Sidorovich AV, Koton MM (1977) Effect of molecular orientation and crystallization on mechanical properties of oriented polypyromellitimides. Polym Sci USSR 19:1297–1304
Shibayev LA, Sazanov YN, Stepanov NG, Bulina TM, Zhukova TI, Koton MM (1982) Mass spectrometric thermal analysis of the cyclodehydration of polyamic acids. Polym Sci USSR 24:2922–2929
Sidorovich AV, Baklagina YG, Mikhailova NV, Prokhorova LK, Schcherbakova LM, Koton MM (1983) Supermolecular-structure formation in films prepared from the dianhydride of 3,3′,4,4′-diphenyloxide tetracarboxylic acid and various diamines. Polym Sci USSR 25:2433–2441
Buckley LJ, Eashoo M (1999) Wet-spinning fiber process providing controlled morphology of the wet-spun fiber. US Patent 5938999
Eashoo M, Shen D, Wu Z, Lee CJ, Harris FW, Cheng SZD (1993) High-performance aromatic polyimide fibres: 2. Thermal mechanical and dynamic properties. Polymer 34:3209–3215
Eashoo M, Wu Z, Zhang A, Shen D, Tse C, Harris FW, Cheng SZD, Gardner KH, Hsiao BS (1994) High performance aromatic polyimide fibers, 3. A polyimide synthesized from 3,3′,4,4′-biphenyltetracarboxylic dianhydride and 2,2′-dimethyl-4,4′-diaminobiphenyl. Macromol Chem Phys 195:2207–2225
Harris FW, Cheng SZD (1995) Process for preparing aromatic polyimide fibers. US Patent 5378420
Hsiao BS, Kreuz JA, Cheng SZD (1996) Crystalline homopolyimides and copolyimides derived from 3,3‘,4,4‘-biphenyltetracarboxylic dianhydride/1,3-bis(4-aminophenoxy) benzene/1,12-dodecanediamine. 2. Crystallization, melting, and morphology. Macromolecules 29:135–142
Kim YH, Harris FW, Cheng SZD (1996) Crystal structure and mechanical properties of ODPA-DMB polyimide fibers. Thermochim Acta 282:411–423
Li F, Fang S, Ge JJ, Honigfort PS, Chen JC, Harris FW, Cheng SZD (1999) Diamine architecture effects on glass transitions, relaxation processes and other material properties in organo-soluble aromatic polyimide films. Polymer 40:4571–4583
Li F, Ge JJ, Honigfort PS, Fang S, Chen JC, Harris FW, Cheng SZD (1999) Dianhydride architectural effects on the relaxation behaviors and thermal and optical properties of organo-soluble aromatic polyimide films. Polymer 40:4987–5002
Li F, Kim KH, Savitski EP, Chen JC, Harris FW, Cheng SZD (1997) Molecular weight and film thickness effects on linear optical anisotropy of 6FDA-PFMB polyimides. Polymer 38:3223–3227
Li W, Wu Z, Jiang H, Eashoo M, Harris FW, Cheng SZD (1996) High-performance aromatic polyimide fibres. J Mater Sci 31:4423–4431. doi:10.1007/BF00356470
Wu TM, Chvalun S, Blackwell J, Cheng SZD, Wu Z, Harris FW (1995) X-ray analysis and molecular modelling of the structure of aromatic copolyimides. Polymer 36:2123–2131
Gao G, Liang D, Liu X, Ye G, Gu Y (2008) Structure and properties of novel PMDA/ODA/PABZ polyimide fibers. Polym Eng Sci 48:912–917
Goel RN, Hepworth A, Deopura BL, Varma IK, Varma DS (1979) Polyimide fibers: structure and morphology. J Appl Polym Sci 23:3541–3552
Weinrotter K, Jeszenszky T, Schmidt H, Baumann S, Kalleitner J (1989) Non-flammable, high-temperature resistant polyimide fibers made by a dry spinning method. US Patent 4801502
Kaneda T, Katsura T, Nakagawa K, Makino H, Horio M (1986) High-strength–high-modulus polyimide fibers I. One-step synthesis of spinnable polyimides. J Appl Polym Sci 32:3133–3149
Kaneda T, Katsura T, Nakagawa K, Makino H, Horio M (1986) High-strength–high-modulus polyimide fibers II. Spinning and properties of fibers. J Appl Polym Sci 32:3151–3176
Chang JJ, Liu WW, Zhang MY, Cao L, Ge QY, Niu HQ, Sui G, Wu DZ (2015) Structures and properties of polyimide fibers containing fluorine groups. RSC Adv 5:71425–71432
Chang JJ, Niu HQ, He M, Sun M, Wu DZ (2015) Structure–property relationship of polyimide fibers containing ether groups. J Appl Polym Sci 132:42474
Chen YT, Zhang QH (2014) Synthesis and properties of polyimides derived from diamine monomer containing bi-benzimidazole unit. J Polym Res 21:9895–9906
Chen YT, Zhang QH (2015) Synthesis, characterization and properties of aromatic copolyimides containing Bi-benzimidazole moiety. J Polym Res 22:78
Dong J, Yin C, Lin J, Zhang D, Zhang Q (2014) Evolution of the microstructure and morphology of polyimide fibers during heat-drawing process. RSC Adv 4:44666–44673
Dong J, Yin C, Zhang Z, Wang X, Li H, Zhang Q (2014) Hydrogen-bonding interactions and molecular packing in polyimide fibers containing benzimidazole units. Macromol Mater Eng 299:1170–1179
Feng Y, Luo LB, Huang JY, Li K, Li BY, Wang HN, Liu XY (2016) Effect of molecular rigidity and hydrogen bond interaction on mechanical properties of polyimide fibers. J Appl Polym Sci 133:43677
Gan F, Dong J, Tan W, Zhang D, Zhao X, Chen X, Zhang Q (2017) Fabrication and characterization of co-polyimide fibers containing pyrimidine units. J Mater Sci 52:9895–9906. doi:10.1007/s10853-017-1099-1
Liu XY, Xu W, Ye GD, Gu Y (2006) Novel aromatic polyimide fiber with biphenyl side-groups: dope synthesis and filament internal morphology control. Polym Eng Sci 46:123–128
Luo LB, Yao J, Wang X, Li K, Huang JY, Li BY, Wang HN, Feng Y, Liu XY (2014) The evolution of macromolecular packing and sudden crystallization in rigid-rod polyimide via effect of multiple H-bonding on charge transfer (CT) interactions. Polymer 55:4258–4269
Luo LB, Zheng YX, Huang JY, Li K, Wang HN, Feng Y, Wang X, Liu XY (2015) High-performance copoly(benzimidazole-benzoxazole-imide) fibers: fabrication, structure, and properties. J Appl Polym Sci 132:42001
Niu HQ, Huang MJ, Qi SL, Han EL, Tian GF, Wang XD, Wu DZ (2013) High-performance copolyimide fibers containing quinazolinone moiety: preparation, structure and properties. Polymer 54:1700–1708
Niu HQ, Qi SL, Han EL, Tian GF, Wang XD, Wu DZ (2012) Fabrication of high-performance copolyimide fibers from 3,3′,4,4′-biphenyltetracarboxylic dianhydride, p-phenylenediamine and 2-(4-aminophenyl)-6-amino-4(3H)-quinazolinone. Mater Lett 89:63–65
Sun M, Chang JJ, Tian GF, Niu HQ, Wu DZ (2016) Preparation of high-performance polyimide fibers containing benzimidazole and benzoxazole units. J Mater Sci 51:2830–2840. doi:10.1007/s10853-015-9591-y
Yang WK, Liu FF, Zhang ES, Qiu XP, Xl Ji (2017) Influence of atmosphere and force during thermal imidization on the structure and properties of BPDA–PDA polyimide fibers. Chem J Chin Univ 38:150–158
Yi XL, Gao YY, Zhang MY, Zhang CB, Wang Q, Liu GM, Dong X, Wu DZ, Men YF, Wang DJ (2017) Tensile modulus enhancement and mechanism of polyimide fibers by post-thermal treatment induced microvoid evolution. Eur Polymer J 91:232–241
Yin CQ, Dong J, Tan WJ, Lin JY, Chen DJ, Zhang QH (2015) Strain-induced crystallization of polyimide fibers containing 2-(4-aminophenyl)-5-aminobenzimidazole moiety. Polymer 75:178–186
Yin CQ, Dong J, Zhang DB, Lin JY, Zhang QH (2015) Enhanced mechanical and hydrophobic properties of polyimide fibers containing benzimidazole and benzoxazole units. Eur Polymer J 67:88–98
Yin CQ, Dong J, Zhang ZX, Zhang QH, Lin JY (2015) Structure and properties of polyimide fibers containing benzimidazole and amide units. J Polym Sci Part B Polym Phys 53:183–191
Yin CQ, Zhang ZX, Dong J, Zhang QH (2015) Structure and properties of aromatic poly(benzimidazole-imide) copolymer fibers. J Appl Polym Sci 132:41474
Zhang MY, Niu HQ, Chang JJ, Ge QY, Cao L, Wu DZ (2015) High-performance fibers based on copolyimides containing benzimidazole and ether moieties: molecular packing, morphology, hydrogen-bonding interactions and properties. Polym Eng Sci 55:2615–2625
Zhang MY, Niu HQ, Lin ZW, Qi SL, Chang JJ, Ge QY, Wu DZ (2015) Preparation of high performance copolyimide fibers via increasing draw ratios. Macromol Mater Eng 300:1096–1107
Angelo RJ (1969) Polyamides and polyimides from diamines containing alkoxy phenol groups. US Patent 3420795
Haller JR (1970) Shaped polyimides and method. US Patent 3494823
Samuel IR, Edgar SC (1968) Formation of polypyromellitimide filaments. US Patent 3415782
Zhang QH, Dai M, Ding MX, Chen DJ, Gao LX (2004) Mechanical properties of BPDA–ODA polyimide fibers. Eur Polymer J 40:2487–2493
Coburn JC, Soper PD, Auman BC (1995) Relaxation behavior of polyimides based on 2,2′-disubstituted benzidines. Macromolecules 28:3253–3260
Comer AC, Kalika DS, Rowe BW, Freeman BD, Paul DR (2009) Dynamic relaxation characteristics of Matrimid® polyimide. Polymer 50:891–897
Saeed MB, Zhan MS (2006) Effects of monomer structure and imidization degree on mechanical properties and viscoelastic behavior of thermoplastic polyimide films. Eur Polymer J 42:1844–1854
Xu YK, Zhan MS, Wang K (2004) Structure and properties of polyimide films during a far-infrared-induced imidization process. J Polym Sci Part B Polym Phys 42:2490–2501
Acknowledgements
The authors are grateful to the fund of the National High Technology Research and Development of China (973 Program) administered by the Ministry of State Science and Technology for their support of this work (Project No. 2014CB643606). Besides, the authors would like to express appreciations to Prof. Er-Qiang Chen and his coworkers at Peking University for their great support and useful discussions on WAXD analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yan, X., Zhang, M., Qi, S. et al. A high-performance aromatic co-polyimide fiber: structure and property relationship during gradient thermal annealing. J Mater Sci 53, 2193–2207 (2018). https://doi.org/10.1007/s10853-017-1552-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1552-1