
Abstract
Strontium (Sr) enhances bone formation both in vitro and in vivo, while it reduces bone resorption. Thus, Sr incorporation in bioactive glass–ceramic scaffolds for bone tissue regeneration could further enhance osteogenesis. The aim of this work was the synthesis, characterization and investigation of the apatite-forming ability in inorganic environment of two sol–gel-derived bioactive Sr-containing glass–ceramic materials with 5 and 10% of SrO. The thermal properties of the synthesized materials were studied using differential thermal analysis (TG–DTA). The apatite-forming ability test was conducted in SBF for various immersion times for both thermally treated and untreated samples. The characterization of the samples before and after immersion in SBF was performed with Fourier transform infrared spectroscopy (FTIR), X-ray powder diffraction (XRD) and scanning electron microscopy with associated energy-dispersive spectroscopy. FTIR spectra revealed that all synthesized glass–ceramic materials presented the characteristic bands of silicate glasses, while XRD identified various crystalline phases, mostly calcium silicates. Strontium is present in the form of strontium silicate in both as-received and thermally treated specimens, and Sr-diopside in the thermally treated specimens. The apatite-forming ability of the glass–ceramic materials was confirmed by the formation of a hydroxyapatite layer after 3 and 5 days of immersion in SBF on the surface of the untreated and thermally treated samples, respectively. The apatite layer, also, became thicker as the immersion time increased.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biomaterials are either natural or artificial materials which can be inserted in biological systems, in order to improve or replace damaged organic tissues or organs [1, 2] causing little or no side effects in the human body [3]. The ability of biomaterials to interact with the tissues is called bioactivity [1]. Due to this property, an exchange of ions occurs between the implanted bioactive materials and the body fluids and as a result a hydroxycarbonate apatite (HCAp) layer is being developed at the interface between them and the surrounding tissues. Through this layer, a strong connection is achieved [4,5,6,7].
Biomaterials, according to their chemical nature, are being classified in ceramics, metals and metal alloys, polymers and composite materials [8]. As ceramics we name all inorganic non-metallic materials which have undergone heat treatment at high temperatures [9]. Any ceramic which presents some kind of biocompatibility is called bioceramic [6]. Since the discovery of Prof. Hench of 45S5 bioactive glass [10, 11], various compositions of bioactive glasses (amorphous solid materials) and glass–ceramics which are placed in the range of bioceramic materials [8] and are suitable for bone tissue engineering applications have been proposed [12]. Many of these glass–ceramics were synthesized with the sol–gel technique. The sol–gel technique is a widely used method for the production of bioactive glass–ceramic materials because of its advantages, mainly the enhanced homogeneity, porosity and bioactive behavior of the synthesized materials and the increased surface area that these materials present [13, 14]. Furthermore, various studies have been reported in which many sol–gel produced materials have the ability to bond to both soft and hard tissues and to promote bone growth due to their increased surface area [15]. The sol–gel route is effectively employed in the development of novel bioceramic scaffolds for bone tissue engineering [16]. The sol–gel process allows the presence of nanopores in the resulting material, which in turn is causing its degradation in a faster rate than melt-derived bioceramics, ultimately resulting in faster HCAp formation [17]. Additionally, several studies report that Si-, Ca-, Mg- and Sr-containing glasses are highly bioactive and could be used for biomedical applications [12, 18, 19]. The controlled release of these active metal ions, at desired dosage levels in the physiological environment, affects positively the behavior of human cells and enhances the therapeutic and stimulant effects of bioactive glasses in osteogenesis, angiogenesis, and antibacterial potential [20,21,22]. Specifically, Sr-containing bioactive glasses and glass–ceramics have attracted the interest of the scientific community because of the beneficial effect of strontium (Sr) on bone metabolism and their improved mechanical properties. The major advantage of Sr is that it reduces bone resorption, while it enhances bone formation both in vitro and in vivo [15, 23], becoming a promising agent for the treatment of osteoporosis [24, 25]. Bioactive Sr-containing glasses have increased extracellular bioactivity and release critical concentrations of the Sr-ions in the dissolution, within the range 1–5 ppm [26, 27]. This release of Sr-ions has been shown to enhance bone cell activity [28].
Although recent research has shifted the emphasis to the importance of incorporating divalent ions such as Mg2+, Zn2+ and Sr2+ in calcium silicate (CS) bioceramics for the enhancement of bone formation and/or regeneration, the synergistic effects of these ions either on the apatite-forming ability or osteogenic capacity have not been thoroughly investigated. To the best of the authors’ knowledge, sol–gel bioactive CS ceramics combining two important trace elements of bone, i.e., Mg2+ and Sr2+, that have proven positive effect on bone metabolism have not been evaluated before. These novel materials could prove beneficial as multi-ion-releasing substances and could be used in many versatile ways such as the synthesis of bioceramic scaffolds or bone cements, aiming to exhibit better biological performance toward bone generation, especially in cases of osteoporotic bone defects where high-quality bone is difficult to regenerate.
In this work, two Sr-containing glass–ceramic materials in the ternary system SiO2–CaO–MgO with different Sr content were synthesized by the sol–gel method [27]. The apatite-forming ability test of glass–ceramics was conducted in SBF for various immersion times (3, 5, 10, 15, 20 and 30 days). The bioactive response of the samples was evaluated using Fourier transform infrared spectroscopy (FTIR), differential thermal and thermogravimetric analysis (TG–DTA), X-ray diffraction analysis (XRD) and scanning electron microscopy with associated energy-dispersive spectroscopic analysis (SEM/EDS).
Materials and methods
Two glass–ceramic materials were prepared, with similar composition nominally SiO2 60, CaO 30, MgO 5, SrO 5 (Sr5) and SiO2 60, CaO 25, MgO 5, SrO 10 (Sr10) in wt%. The glasses were synthesized by the sol–gel technique as described in the literature [29,30,31]. In detail, the two glasses were synthesized by mixing 29 ml of DI H2O, 4.8 ml of 2 N HNO3 and 30 ml of tetraethyl orthosilicate (TEOS) in a beaker. A magnet was added to the solution, and the beaker was placed on stirrer at 400 rpm, until the solution was hydrolyzed [32]. Afterward, the amounts of Ca(NO3)2·4H2O, Mg(NO3)2·6H2O and Sr(NO3)2 were accordingly calculated and added in the composition. The solution was left to stir for 50 min at room temperature. Aging was achieved at 60 °C for 55 h, and afterward, for drying, the materials were heated gradually at 60, 90 and 120 °C for 69 h, in the presence of moisture. Then the materials were transferred to a platinum capsule (Pt), undergoing heat treatment at 400, 600 and 700 °C, to achieve the final stage of chemical stabilization, that is to allow nitrate elimination and the final densification of the resulted materials.
At this point, the two Sr-containing materials (Sr5 and Sr10) were categorized in two sub-groups each. One sub-group included the materials that were kept as-received after chemical stabilization (Sr5u and Sr10u) and the second group the materials that were subjected to subsequent thermal treatment (Sr5t and Sr10t) according to the results of the thermal analysis. Thermal treatment of both groups was performed from room temperature (18 °C) to 892 and 903 °C for the Sr5 and the Sr10, respectively, with a heating rate of 10 °C/min. At these temperatures, they remained for 10 min and then they were cooled in room temperature.
All glasses were characterized by differential thermal and thermogravimetric analysis, performed with a Setaram thermogravimetric–differential thermal analyzer SETSYS 16/18-TG–DTA, dynamic conditions with heating rates of 10 °C/min—in nitrogen atmosphere—and furnace cooling. The samples were placed in alumina crucibles, and heating was performed up to 1350 °C. The sequential sintering temperature was chosen based on the maximum crystallization of the materials according to the thermogram.
The obtained powders were placed into a metal die and pulverized in vacuum under pressure of <10 tons. Thereafter, the powders were transferred to a ball milling (S100, Retsch GmbH, Germany) and were ground for 30 min at a rate of 300 rpm. The milled powders with a grain size of <40 mm were collected through a mesh sieve.
The apatite-forming ability test of glass–ceramics was conducted in c-SBF (pH 7.40) at 37 °C with a ratio of 1.5 mg/ml [33] for various immersion times (3, 5, 10, 20 and 30 days). The SBF solution was prepared as described by Kokubo et al. [34] and had similar ion concentrations to those of human blood plasma. Then the samples were removed from the SBF solution and they were rinsed with ethanol and distilled water, dried at 30–40 °C and stored in airtight containers.
Before and after the heat treatment, as well as the apatite-forming ability test, the crystal structure of the material was investigated by Fourier transform infrared spectroscopy (FTIR), X-ray diffractometry (XRD) and scanning electron microscopy with associated energy-dispersive spectroscopy (SEM/EDS). FTIR transmittance spectra were obtained using a Perkin-Elmer Spectrometer Spectrum 1000 in MIR region (4000–400 cm−1) with a resolution 4 cm−1, by the KBr pellet technique. XRD measurements were carried out using a Philips (PW1710) diffractometer with Ni-filtered CuKa wave radiation. The counting statistics of the XRD study were: step size: 0.02° 2θ, start angle: 5°, end angle: 75° and the time per step: 1.5 s. Recognition and identification of crystalline phases was based on the database PDF-4+ (2009) of the ICDD. The quantitative estimation of the minerals was based on counts of specific reflections, taking under consideration the density and the mass absorption coefficient of minerals identified for CuKa radiation. Corrections were made using Rietveld refinement. The proportion of amorphous material was calculated according to the methodology described by Kantiranis et al. [35]. The synthesized materials were powdered. The topographical evaluation and elemental analysis of the samples before and after immersion in SBF were performed by a scanning electron microscope (J.S.M. 840A; JEOL, Tokyo, Japan) with associated energy-dispersive X-ray spectrometer (OXFORD, INCA X-Stream 2, UK) (SEM/EDS).
Results and discussion
Differential thermal and thermogravimetric analysis
Figure 1 shows the mass loss and the heat flow curves of the Sr-containing materials. For both synthesized materials, mass reduction in TG curves took place in certain individual steps. The primary two mass loss steps concern water elimination, due to the evaporation either of adsorbed water or of resulting water during the polycondensation reaction of silanol [36, 37]. The mass loss at the range of 420–675 °C is associated with the thermal decomposition of portlandite, while the most pronounced mass reduction occurred in the temperature range of 675–920 °C. Specifically, for Sr5 specimen the beginning of that step occurred at 650 °C, resulting in a 0.8% mass loss that was completed at 880 °C. For the Sr10 material, respectively, the beginning of the mass reduction appeared at 687 °C and its completion occurred at 920 °C, with a total mass loss of 2%. The aforementioned mass losses were attributed to the loss of CO2, probably due to the partial decomposition of strontianite, the presence of which was confirmed by XRD [38]. The total mass loss of both materials was well under 4%.

Heat flow and mass loss curves for a Sr5 and b Sr10 glass–ceramic materials
The heat flow curves of both specimens exhibited two endothermic peaks up to 430 °C, which were attributed to the aforementioned processes of water evaporation. Afterward, there was a small detectable endothermic peak (754 °C) for both materials, which was probably caused by a glass transition process. The heat flow curve of Sr5 presented a crystallization peak at 892 °C, whereas the Sr10 curve presented a peak at 903 °C and a smaller one at and 896 °C (embedded image in Fig. 1b). This difference in the thermal behavior of the Sr-containing materials has been associated with the amount of Sr in the glass network. As reported by Massera and Hupa [39] when up to 5 mol% of SrO is added in order to substitute the CaO content of a bioactive glass, the main crystallization peak shifts to lower temperatures. However, when the amount of SrO is higher than 5 mol%, the main crystallization peak is slightly shifted to higher temperatures, while a second crystallization peak is formed quite near, in slightly lower temperatures. Finally, the melting process took place in the temperature range of 1235 °C up to the end of the obtained curves, consisting of two distinct peaks for both materials, attributed to the melting of different crystalline phases [40]. More specifically, the two discrete endothermic peaks of Sr5 sample occurred at 1294 °C and 1347 °C, while those of Sr10 specimen appeared at 1329 and 1332 °C. The crystallization peaks at 892 and 903 °C for each material were selected as the sintering temperature.
FTIR analysis of the synthesized materials
Figure 2 shows FTIR spectra of the synthesized powders. FTIR spectroscopy revealed that all synthesized glass–ceramic materials presented the characteristic bands of silicate glasses, those being the broad peak at 900–1200 cm−1 and the peak at 400–580 cm−1 [41]. These peaks are attributed to the asymmetric stretching vibration of Si–O–Si and the bending vibration of the Si–O–Si bonds, respectively [42]. Furthermore, the peak at 1646 cm−1 indicates the presence of absorbed water on the surface of the samples [43]. The FTIR spectra of untreated specimens present a sharp peak at 3643 cm−1 attributed to Ca(OH)2, while the broad band at ~1460 cm−1 along with the weak band at 859 cm−1 indicate the presence of SrCO3. Moreover a small quantity of calcium silicate crystalline phases is observed, especially for the Sr10u sample. Comparing the spectra of treated to untreated samples, a significant reduction of Ca(OH)2 and SrCO3 is observed. Also, the new peaks that emerge indicate the increase in crystalline phases such us wollastonite and Sr-diopside.

FTIR spectra of the synthesized materials
SEM/EDS analysis of the synthesized materials
SEM microphotographs of the synthesized materials (Fig. 3) revealed irregular-shaped grains with a grain size under 40 μm. EDS analysis did not reveal any significant alterations in elemental composition between untreated and thermally treated samples, while higher amounts of Sr were detected in the Sr10u and Sr10t samples as was expected.

SEM microphotographs of the synthesized powders with indicative EDS spectra from spot analysis
XRD analysis of the synthesized materials
The XRD patterns of the synthesized powders are shown in Fig. 4. The results are in accordance with the FTIR findings, while significant differences were observed among the crystalline phases of the untreated and treated samples, which are shown briefly in Table 1.

XRD patterns of the synthesized powders before and after immersion in SBF solution
As shown by XRD analysis, both groups present large amounts of crystalline phases which increase after thermal treatment. The main phases are calcium silicates, as was expected, while Sr is present in the form of Sr silicate in both the as-received materials and the thermally treated specimens, and Sr-diopside in the thermally treated specimens. Pyroxene group is a large group of inosilicate (chain silicate) minerals with the general formula XYZ2O6, where X can be one of the following elements: Mg2+, Fe2+, Mn2+, Li+, Ca2+, Na+, Y: Al3+, Fe3+, Cr3+, Cr3+, Ti4+, Mg2+, Fe2+, Mn2+ and Z: Si, Al3+, Fe3+ [44]. Although diopside (CaMgSi2O6) is the most common form of pyroxene in Mg-based glass–ceramics, in both thermally treated groups, Sr-diopside has been identified in small percentages, suggesting the substitution of Ca2+ with Sr2+ in its structure. This substitution is common, as Sr2+ and Ca2+ have similar ionic radii of 1.18 and 1.00 Å, respectively [45], a difference that allows Sr to be substituted for Ca in bioactive glasses [46]. However, as the ionic radius of Sr is slightly higher, it leads to an expansion and subsequent disruption of the glass network, altering the nucleation mechanisms facilitating the crystallization process [47].
FTIR analysis of the synthesized materials after immersion in SBF
The FTIR spectra (Fig. 5) reveal the precipitation of apatite on the surface of all samples after 5 days of immersion in SBF as verified by the strong band at ~1035 cm−1 which is assigned to the P–O stretching vibration and the appearance of the two bands at 610–600 and 560–550 cm−1 assigned to the P–O bending vibration of HAp [43, 48,49,50]. The double peak of the P–O bending vibration of HAp appears first in the spectra of untreated specimens, after 3 days of immersion, and with 2 days delay in the spectra of the thermally treated specimens. In a recent paper of Goudouri et al. [51], a Sr-free sol–gel-derived glass in the system 60%SiO2–30%CaO–10%Mg0 thermally treated 20–40 °C below its crystallization temperature, developed amorphous Ca–P spheres after 3 days in SBF. Thus, Sr substitution in the glass–ceramics of the present study, did not exert any negative effect on the apatite-forming ability of the synthesized materials, but instead resulted in the formation of crystalline HAp even after 5 days in SBF. The peak at 800 cm−1, which emerges after 3 days in SBF, corresponds to the Si–O–Si stretching vibration due to the polycondensation of silanols (SiOH + SiOH → Si–0–Si) [7]. This peak disappears in the spectra of both Sr5u and Sr10u after 30 and 20 days, respectively, while it still remains in the spectra of both Sr5t and Sr10t even after 30 days in SBF. For the untreated samples, it seems that apatite formation starts earlier on the Sr10u samples with the higher amount of Sr, which can be explained by the less coherent glass network due to the higher amount of Sr substitution of Ca and by the higher MgO/CaO ratio which enhances the surface reactivity of bioactive glasses [51,52,53]. Comparing Sr5 to Sr10 samples, there is no significant difference concerning the time for apatite precipitation. Comparing untreated to treated samples, a delay in the growth of apatite is observed for the treated samples, as shown by the presence of the peak at 800 cm−1 even after 30 days in SBF and the presence of the peak at 465 cm−1 attributed to the Si–O–Si bending vibration from the substrate silica network. Higher ion solubility for the untreated samples due to their less crystallinity (higher amount of amorphous and lower amount of calcium silicates as shown later with XRD) may explain this difference in apatite-forming ability. The peak at 870 cm−1 assigned to the C–O out-of-plane bending vibration of the carbonate group [43, 50] is gradually increased during the immersion in SBF. Although this peak has been assigned to the HCAp formation, XRD analysis failed to reveal the formation of carbonated apatite. As shown later, other carbonated crystalline phases are developed during the immersion in SBF which justify the increase in the intensity of this peak [54].

FTIR spectra of the untreated and thermally treated powders reacted in SBF for various days of immersion for each sample group. a Sr5u, b Sr10u, c Sr5t and d Sr10t
Summarizing, after 5 days of immersion in SBF an apatite layer is developed on the surface of all samples, which becomes thicker with time, as verified by the sharper and more intense peaks of apatite, especially on the untreated specimens.
XRD analysis of the synthesized materials after immersion in SBF
The percentages of the identified crystalline phases by XRD are shown in Table 2 and the respective patterns in Fig. 4. The intensity of the amorphous phase is increased with immersion time firstly due to the decrease of the percentages of the phases of the starting materials. Secondly the apatite-forming ability of a material, while it is immersed in SBF, constitutes an ongoing procedure. Thus, the increase in amorphous content is inherently dependent on apatite precipitation; why the amorphous phase is the precursor for that procedure. Also the precipitation of apatite on the surface of all samples after 5 days of immersion in SBF is verified. These data are consistent with the results of the FTIR spectra. A delay in the growth of apatite is observed concerning the Sr10t samples, which was expected due to its increased crystallinity. However, after 20 days the percentages of apatite for both materials either untreated or not are comparable. The developed apatite layer on the surface of all samples becomes thicker with time, as verified by the higher amounts of HAp calculated by the XRD. An interesting finding was observed by the XRD analysis of the treated specimens, as minor amounts of vaterite were recorded after immersion in SBF. Vaterite is one of the three predominant anhydrous calcium carbonate mineral phases of CaCO3, with the other two being calcite and aragonite. From these polymorphs calcite is the more stable, while vaterite is the most metastable phase, which rapidly converts to calcite in wet environment. According to the Ostwald’s rule of stages [55], the least stable phase crystallizes first and then transforms to the more stable polymorphs. Consequently vaterite, as the least stable polymorph of CaCO3, is rarely observed without being intentionally stabilized. In this study, its presence can be justified by factors such as high supersaturation level, alkaline pH, and body temperature (37 °C), conditions that have been reported to favor the formation of the vaterite phase [56, 57]. However, vaterite was not observed on the surface of untreated specimens. This can be explained by the high amount of calcium silicates present on the thermally treated specimens. Calcium carbonate formation in the presence of various silicates and oxides shows a non-predicted behavior, as the presence of the tetrahedral silicate ion in the flat carbonate lattice prohibits the organization of the carbonate into crystalline polymorphs and favors the kinetic stabilization of vaterite [58,59,60]. In this study, vaterite crystallization may have been favored by the high amount of silica available on the surface of the thermally treated glass–ceramics, in combination with other factors as well, such as temperature and pH of the solution, and the presence of Mg2+ that has been found to suppress the transformation of vaterite by inhibiting the growth of the most stable phase of CaCO3 calcite [61, 62]. Vaterite occurs as sub-μm-sized spherical particles, and it has been recently proposed along with aragonite, as additive in resorbable calcium phosphate cements for bone tissue engineering, as it can develop apatitic CaP globules between 72 and 96 h of immersion in the Lac-SBF solution at 37 °C [63] (Fig. 4).
SEM/EDS analysis of the synthesized materials after immersion in SBF
The SEM micrographs of the samples are shown in Fig. 6. After 5 days of immersion in SBF, the SEM micrographs revealed changes in the morphology of the surface and the appearance of small agglomerated apatite spheres. After 10 days, the agglomerates start to grow, while the results of the Ca/P ratio ranging from 1.6 to 1.8 suggest HAp precipitation.

SEM microphotographs of the specimens after 5, 10, 20 and 30 days in SBF
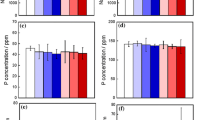
After 30 days in SBF, typical Ca–P EDS spectra were received in all cases of untreated samples with a Ca/P ratio around 1.7 (Fig. 7). In the case of Sr5t and Sr10t and at all immersion times, EDS analysis detected high amounts of calcium along with silicon (Fig. 7, Sr5t, spectrum 2/Sr10t, spectrum 4), due to the presence of calcium silicates on the surface of the starting materials, as detected by XRD. This excess of calcium on the surface of the specimens along with the carbonate ions from SBF allows the formation of vaterite, which has been reported to grow in the presence of calcium silicates as reported above.

Backscattered microphotographs of the specimens after 30 days in SBF and respective EDS analysis
Conclusions
In this study, the successful synthesis of two Sr-containing calcium silicate glass–ceramics was performed through the sol–gel method. Strontium incorporation resulted in the formation of crystalline apatite after 3 and 5 days of immersion in SBF on the surface of the untreated and thermally treated samples, respectively. Due to their high amount of amorphous glassy phase and their enhanced apatite-forming ability even after sintering at high temperatures, these novel glass–ceramics can be suggested for the synthesis of Sr-containing/releasing scaffolds for bone tissue repair or engineering. Nevertheless, in order to examine the kinetics of Sr release and to provide insights of its beneficial effect on cell attachment, proliferation and differentiation, further work is currently contacted.
References
Ratner BD, Hoffman AS, Schoen FJ, Lemons JE (2013) Biomaterials science: an introduction to materials in medicine, 3rd edn. Elsevier Inc., Oxford
Ong KL, Lovald S, Black J (2014) Biological performance of materials in research and practice. Taylor and Francis Group, Boca Raton
Doremus RH (1992) Review bioceramics. J Mater Sci 27:285–297. doi:10.1007/BF00543915
Kokubo T (1997) Novel bioactive materials. An Quím Inter Edn 93(1):549–555
Hench LL, Kokubo T (1988) Handbook of biomaterial properties. Chapman and Hall, London by Black J and Hastings G
Hench LL (1991) Bioceramics: from concept to clinic. J Am Ceram Soc 74(7):1487–1510
Peitl Filho O, LaTorre GP, Hench LL (1996) Effect of crystallization on apatite-layer formation of bioactive glass 45S5. J Biomed Mater Res 30(4):509–514
Vallet-Regi M (2001) Ceramics for medical applications. J Chem Soc Dalton Trans 2:97–108
Caruta BM (2006) Ceramics and composite materials: new research. Nova Science Publishers Inc., New York
Hench LL, Splinter RJ, Allen WC, Greenlee TK Jr (1971) Bonding mechanisms at the interface of ceramic prosthetic materials. J Biomed Mater Res 2:117–141
Greenlee TK Jr, Beckham CA, Crebo AR, Malmborg JC (1972) Glass ceramic bone implants. J Biomed Mater Res 6:235–244
Hoppe A, Güldal NS, Boccaccini AR (2011) A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials 32:2757–2774
Hench LL, Thompson I (2010) Twenty-first century challenges for biomaterials. J R Soc Interface 7:379–391
Hench LL, Jones JR, Sepulveda P (2005) Bioactive materials for tissue engineering scaffolds. Imperial College Press, London
Marie PJ, Ammann P, Boivin G, Rey C (2001) Mechanisms of action and therapeutic potential of strontium in bone. Calcif Tissue Int 69(3):121–129
Baino F, Fiorilli S, Vitale-Brovarone C (2016) Bioactive glass-based materials with hierarchical porosity for medical applications: review of recent advances. Acta Biomater 42:18–32
Rahaman MN, Day DE, Bal BS, Fu Q, Jung SB, Bonewald LF, Tomsia AP (2011) Bioactive glass in tissue engineering. Acta Biomater 7(6):2355–2373
Wu C, Chang J (2006) A novel akermanite bioceramic: preparation and characteristics. J Biomater Appl 21:119–129
Xynos ID, Edgar AJ, Buttery LDK, Hench LL, Polak M (2001) Gene-expression profiling of human osteoblasts following treatment with the ionic products of Bioglass® 45S5 dissolution. J Biomed Mater Res 55(2):151–157
Jones JR (2012) Review of bioactive glass: from hench to hybrids. Acta Biomater 9:4457–4486
Baino F, Novajra G, Miguez-Pacheco V, Boccaccini AR, Vitale-Brovarone C (2016) Bioactive glasses: Special applications outside the skeletal system. J NonCrystal Sol 432(Part A):15–30
Wu C, Chang J (2014) Multifunctional mesoporous bioactive glasses for effective delivery of therapeutic ions and drug/growth factors. J Control Release 193:282–295
Marie PJ (2006) Strontium ranelate: a physiological approach for optimizing bone formation and resorption. Bone 38(2, Suppl. 1):10–14
Meunier PJ, Slosman DO, Delmas PD, Sebert JL, Brandi ML, Albanese C, Lorenc R, Pors-Nielsen S, De Vernejoul MC, Roces A, Reginster JY (2002) Strontium ranelate: dose-dependent effects in established postmenopausal vertebral osteoporosis—a 2-year randomized placebo controlled trial. J Clin Endocrinol Metab 87(5):2060–2066
Bonnelye E, Chabadel A, Saltel F, Jurdic P (2008) Dual effect of strontium ranelate: stimulation of osteoblast differentiation and inhibition of osteoclast formation and resorption in vitro. Bone 42:129–138
Hesaraki S, Alizadeh M, Nazarian H, Sharifi D (2010) Physico-chemical and in vitro biological evaluation of strontium/calcium silicophosphate glass. J Mater Sci Mater Med 21(2):695–705. doi:10.1007/s10856-009-3920-0
Lao J, Jallot E, Nedelec JM (2008) Strontium-delivering glasses with enhanced bioactivity: a new biomaterial for antiosteoporotic applications? Chem Mater 20(15):4969–4973
Gentleman E, Fredholm YC, Jell G, Lotfibakhshaiesh N, O’Donnell MD, Hill RG, Stevens MM (2010) The effects of strontium-substituted bioactive glasses on osteoblasts and osteoclasts in vitro. Biomaterials 31(14):3949–3956
Zhong J, Greenspan DC (2000) Processing and properties of sol–gel bioactive glasses. J Biomed Mater Res 53(6):694–701
Wu C, Chang J (2004) Synthesis and apatite-formation ability of akermanite. Mater Lett 58(19):2415–2417
Wu C, Chang J, Zhai W, Ni S, Wang J (2006) Porous akermanite scaffolds for bone tissue engineering: preparation, characterization, and in vitro studies. J Biomed Mater Res 78(1):47–55
Brinker CJ (1988) Hydrolysis and condensation of silicates: effects on structure. J Non-Cryst Solids 100:31–50
Wu C, Chang J (2007) Degradation, bioactivity, and cytocompatibility of diopside, akermanite, and bredigite ceramics. J Biomed Mater Res 83B:153–160
Kokubo T, Kushitani H, Sakka S, Kitsugi T, Yamamuro T (1990) Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W. J Biomed Mater Res 24:721–734
Kantiranis N, Stergiou A, Filippidis A, Drakoulis A (2004) Calculation of the percentage of amorphous material using PXRD patterns. Bull Geol Soc Greece 36:446–453
Chatzistavrou X, Zorba T, Chrissafis K, Kaimakamis G, Kontonasaki E, Koidis P, Paraskevopoulos KM (2006) Influence of particle size on the crystallization process and the bioactive behavior of a bioactive glass system. J Therm Anal Calorim 85(2):253–259
Bizari D, Rabiee M, Moztarzadeh F, Tahiriri M, Alavi SH, Masael R (2013) Synthesis, characterization and biological evaluation of sol-gel derived nanomaterial in the ternary system 64%SiO2–31%CaO–5%P2O5 as a bioactive glass: in vitro study. Ceram Silikáty 57(3):201–209
Frost R, Hales M, Martens W (2009) Thermogravimetric analysis of selected group(II) carbonate minerals—implication for the geosequestration of greenhouse gases. J Therm Anal Calorim 95(3):999–1005
Massera J, Petit L, Cardinal T, Videau JJ, Hupa M, Hupa L (2013) Thermal properties and surface reactivity in simulated body fluid of new strontium ion-containing phosphate glasses. J Mater Sci Mater Med 24(6):1407–1416. doi:10.1007/s10856-013-4910-9
Lefebvre L, Chevalier J, Gremillard L, Zenati R, Thollet G, Bernache-Assolant D, Govin A (2007) Structural transformations of bioactive glass 45S5 with thermal treatments. Acta Mater 55:3305–3313
Goudouri OM, Kontonasaki E, Kantiranis N, Chatzistavrou X, Papadopoulou L, Koidis P, Paraskevopoulos KM (2009) Investigation of the bioactivity of dental ceramic/bioactive glass composites prepared by the sol gel route. Key Eng Mater 396–398:119–122
Lenza RFS, Vasconcelos WL (2001) Preparation of silica by sol-gel method using formamide. Mater Res 4(3):189–194
Farmer VC (1973) Chapter 15 the layer silicates. In: Farmer VC (ed) The infrared spectra of minerals. Mineralogical Society, London, pp 352–353
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A 32(5):751–767. doi:10.1107/S0567739476001551
Martin RA, Twyman HL, Rees GJ, Barney ER, Moss RM, Smith JM, Hill RG, Cibin G, Charpentier T, Smith ME, Hanna JV, Newport RJ (2012) An examination of the calcium and strontium site distribution in bioactive glasses through isomorphic neutron diffraction, X-ray diffraction, EXAFS and multinuclear solid state NMR. J Mater Chem 22:22.212–22.223
Hill RG, Stamboulis A, Law RV, Clifford A, Towler MR, Crowley C (2004) The influence of strontium substitution in fluoroapatite glasses and glass–ceramics. J Non-Cryst Solids 336(3):223–229
Ogino M, Ohuchi F, Hench LL (1980) Compositional dependence of the formation of calcium phosphate films on bioglass. J Biom Mater Res 14(1):55–64
Filgueiras MR, La Torre G, Hench LL (1993) Solution effects on the surface reactions of a bioactive glass. J Biomed Mater Res 27(4):445–453
Smith B (1998) Infrared spectra interpretation: a systematic approach. CRC Press, Florida
Goudouri OM, Kontonasaki E, Chrissafis K, Zinn K, Hoppe A, Detsch R, Paraskevopoulos KM, Boccaccini AR (2014) Towards the synthesis of an Mg-containing silicate glass–ceramic to be used as a scaffold for cementum/alveolar bone regeneration. Ceram Int 40(10B):16287–16298
Diba M, Tapia F, Boccaccini AR, Strobel LA (2012) Magnesium-containing bioactive glasses for biomedical applications. Int J Appl Glass Sci 3(3):221–253. doi:10.1111/j.2041-1294.2012.00095.x
Oliveira J, Correia R, Fernandes M, Rocha J (2000) Influence of the CaO/MgO ratio on the structure of phase-separated glasses: a solid state 29Si and 31P MAS NMR study. J Non Cryst Solids 265:221–229. doi:10.1016/S0022-3093(99)00957-6
Fredholm YC, Karpukhina N, Brauer DS, Jones JR, Law RV, Hill RG (2012) Influence of strontium for calcium substitution in bioactive glasses on degradation, ion release and apatite formation. J R Soc Interface 9(70):880–889. doi:10.1098/rsif.2011.0387
Ostwald W (1897) The formation and changes of solids. Z Phys Chem 22:289–330
Zhou GT, Yao QZ, Fu SQ, Guan YB (2010) Controlled crystallization of unstable vaterite with distinct morphologies and their polymorphic transition to stable calcite. Eur J Miner 22(2):259–269. doi:10.1127/0935-1221/2009/0022-2008
Ogino T, Suzuki T, Sawada K (1987) The formation and transformation mechanism of calcium carbonate in water. Geochim Cosmochim Acta 51(10):2757–2767
Lin Y, Hu Q, Chen J, Ji J, Teng HH (2009) Formation of metastable CaCO3 polymorphs in the presence of oxides and silicates. Cryst Growth Des 9(11):4634–4641. doi:10.1021/cg900085e
Gal A, Weiner S, Addadi L (2010) The stabilizing effect of silicate on biogenic and synthetic amorphous calcium carbonate. J Am Chem Soc 132(38):13208–13211. doi:10.1021/ja106883c
Kellermeier M, Glaab F, Klein R, Melero-García E, Kunz W, García-Ruiz JM (2013) The effect of silica on polymorphic precipitation of calcium carbonate: an on-line energy-dispersive X-ray diffraction (EDXRD) study. Nanoscale 5(15):7054–7065. doi:10.1039/c3nr00301a
Kitamura M (2001) Crystallization and transformation mechanism of calcium carbonate polymorphs and the effect of magnesium ion. J Colloid Interface Sci 236(2):318–327
Nebel H, Epple M (2008) Continuous preparation of calcite, aragonite and vaterite, and of magnesium substituted amorphous calcium carbonate (Mg-ACC). Zeitschrift für anorganische und allgemeine Chemie 634(8):1439–1443. doi:10.1002/zaac.200800134
Lee JH, Madden AS, Kriven WM, Tas AC (2014) Synthetic aragonite (CaCO3) as a potential additive in calcium phosphate cements: evaluation in tris-free SBF at 37 °C. J Am Ceram Soc 97(10):3052–3061. doi:10.1111/jace.13124
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Dessou, N.S., Theodorou, G.S., Kantiranis, N. et al. Influence of strontium for calcium substitution on the glass–ceramic network and biomimetic behavior in the ternary system SiO2–CaO–MgO. J Mater Sci 52, 8871–8885 (2017). https://doi.org/10.1007/s10853-017-0914-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-0914-z