
Abstract
One-dimensional SiO2@SiC shell–core nanowire was fabricated on a SiC substrate without any catalyst using a laser sintering method. Based on a solid–liquid–vapor–solid (SLVS) in situ growth mechanism, the as-synthesized SiO2@SiC nanowires of about 60 nm diameter with a shell thickness of about 13 nm were grown. Varying the power density of laser sintering allowed the final SiC particle morphology to be controlled from grainy to whisker-like. High power density or high temperature benefited the nucleation probability of SiC whiskers. The isothermal oxidation test showed that the SiO2@SiC nanowires have good thermodynamic stability at high temperature. The photoluminescence (PL) property of SiO2@SiC nanowire was also investigated. The observed emission peaks were composed of an UV peak centered at 365 nm (3.40 eV) and a blue peak centered at 453 nm (2.74 eV). The theory behind the PL property and the growth mechanism of the SiO2@SiC nanowire were also discussed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Compared with SiC bulk materials, SiC nanowire has higher energy band gap, higher critical breakdown electric field and thermal conductivity, smaller dielectric constant, and good mechanical properties. Therefore, SiC nanowire has become the ideal material for preparing high-frequency, high-powered, low-energy consumption, and high-temperature-resistant devices [1–3].
SiC nanowire is also considered as a suitable short-wavelength light-emitting material. Its application in light-emitting and photoelectronic devices is also a cause for concern. SiO2 itself has excellent mechanical, chemical, and electrical properties, due to which it can be used as the buffer layer and the barrier insulating material in semiconductors and integrated circuit devices. Previous studies have shown that SiC@SiO2 core–shell nanowires possess excellent luminescence property due to their novel structure composed of SiC nanorod core and SiO2 nanotube shell [4–6]. Li et al. and Choi et al. attributed the luminescence phenomenon of SiC@SiO2 to the synergy of oxygen vacancy of SiO2 layer and the size effect of SiC nanowires [7, 8].
To date, SiC@SiO2 core–shell nanostructure has been produced using many methods such as template growth method [9, 10], carbothermal reduction method [11–13], chemical vapor reaction method [5, 14, 15], thermal evaporation [16], and arc-discharge method [6, 17]. However, most of these reported production methods suffered from the assistance of catalyst, complex process, and further purification. In recent years, laser sintering has been employed to fabricate various high-performance ceramics [18–20]. Laser sintering is a rapid processing technology in which laser is used as Manufacturing Energy. Laser sintering is characterized by non-contact, non-polluting, high energy flow and superfast temperature, which is contributed to saving time and improving efficiency. This technology also reduces the risk of contamination with high heating and cooling rates [21]. Moreover, some ceramics with different physical properties [18, 22] can be obtained using laser sintering when compared with conventional sintering techniques. The authors attributed this behavior to the stabilization of a high-temperature phase at room temperature. In this work, laser sintering was employed to prepare SiO2@SiC shell–core nanowire in situ. SiC microparticles were chosen as raw materials without any catalyst. Crystal phase, microstructure, chemical composition, anti-oxidation performance, and photoluminescence (PL) properties of the as-prepared nanowires were investigated. In addition, the theory behind PL and the growth mechanism of SiO2@SiC nanowire were discussed.
Experimental
SiC powders (Sinopharm Chemical Reagent Co. Ltd., Shanghai, China, 99.9% purity) with an average particle size of 1.5 μm were used as the starting materials. The powders were mixed with a binder solution of PVA and uniaxially compacted at 40 MPa into disks with a diameter of 4 mm and a thickness of 1.5 mm.
Sintering was performed using a CO2 laser (HGL-HL-5000) in continuous-wave mode as the heating source. The diameter of laser beam was about 4 mm, which was directed and held at the center of the sample throughout the sintering process. The samples were irradiated by laser beam with the power densities of 4.3 ± 0.1 W/mm2 (1300 ± 15 °C) and 5.9 ± 0.1 W/mm2 (1500 ± 15 °C) for 30 s, respectively. The temperature was measured using a type S thermocouple placed at the center of the sample surface.
Phases and crystalline structures were characterized by X-ray diffraction (XRD, Bruker D8 Advance, Bruker, USA) using Cu K α radiation. Microstructure and morphology of the as-synthesized samples were examined by a field emission scanning electron microscope (FESEM, Nova Nano SEM450, FEI, USA), equipped with an energy-dispersive X-ray spectroscope (EDS, INCA, UK) and a transmission electron microscope (TEM, 2000F, JEOL Ltd., Japan). XPS spectra were measured by a photoelectron spectrometer (XPS, Axis Ultra DLD, Shimadzu, Japan) with monochromatized Al K α radiation. Measurements were done with take-off angle normal to the surface. The oxidation tests were carried out in an electric furnace in air with a heating rate of 5 °C/min at a constant temperature ranging between 1200 and 1600 °C for 3 h. The weight changes by the oxidation were recorded by an electric balance with a resolution of 0.1 mg. PL spectra at room temperature were recorded by a fluorescence spectrophotometer (F-4610, Hitachi, Japan).
Results
The XRD patterns of the as-synthesized samples sintered at 1300 and 1500 °C are presented in Fig. 1. Phase analysis confirmed that SiC phase existed in all the samples sintered at 1300 and 1500 °C. The SiC phase (PDF#29-1129) can be indexed as the (111), (200), (220), (311), and (222) crystal planes. Compared with the samples sintered at 1300 °C, the intensity of SiC diffraction peak significantly increased and Si phase (PDF#27-1402) that was indexed as the (111), (220), and (311) crystal planes only appeared in the samples sintered at 1500 °C. However, no obvious SiO2 phase was detected in all the obtained samples.

XRD patterns of the as-synthesized samples
The particle size of SiC powders used as the starting materials was confirmed by optical microscopic images shown in Fig. 2. The average particle size was determined to be between 1 and 2 μm for the SiC powders. It can be seen that the particle shape of SiC raw materials is irregular.

Morphology of SiC powders used as the starting materials
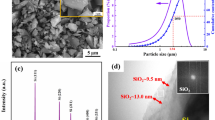
Figure 3 shows the FESEM images and the EDS patterns of the as-synthesized samples sintered using laser sintering at 1300 and 1500 °C. Regular spherical particles were observed for the samples sintered at 1300 °C, which is shown in Fig. 3a. The high-magnification FESEM image of the as-synthesized samples sintered at 1300 °C is displayed in Fig. 3b. The mean grain size of the samples sintered at 1300 °C was measured to be about 250 nm. From the EDS spectra of the samples sintered at 1300 °C shown in Fig. 3c, it can be inferred that the chemical composition consisted mainly of SiO2 along with a little SiC content. In contrast to the morphology of the samples sintered at 1300 °C, a large number of whiskers were formed in the samples sintered at 1500 °C (Fig. 3d). From the high-magnification FESEM image of the samples sintered at 1500 °C (Fig. 3e), it is found that the mean whisker diameter was about 60 nm. From the EDS spectra of the as-synthesized samples sintered at 1500 °C (Fig. 3f), it can be inferred that the chemical composition consisted mainly of SiC and SiO2.

a Low-magnification and b high-magnification FESEM images of the as-synthesized samples sintered at 1300 °C and c mapping EDS patterns of spectrum 1, showing the distribution of the elements (Si, O, and C). d Low-magnification and e high-magnification FESEM images of the as-synthesized samples sintered at 1500 °C and f mapping EDS patterns of spectrum 2, showing the distribution of the elements (Si, C, and O)
Figure 4 shows the TEM images and the EDS spectrum of the as-synthesized sample sintered at 1500 °C. The TEM images exhibited a typical shell–core nanowire structure with a diameter of about 50 nm and a shell thickness of about 13 nm. The EDS spectrum was detected in the position of core. It was found that the main elements were C and Si along with a little O content. This indicated that the chemical composition consisted mainly of SiC.

a Low-magnification and c high-magnification TEM images of the as-synthesized sample sintered at 1500 °C and b EDS spectrum of spectrum 1, showing the distribution of the elements (C, Si, and O)
In order to identify the surface or shell composition of core–shell nanowires, XPS spectrum of the as-synthesized samples sintered at 1500 °C can be used (Fig. 5). As it can be seen, the photoelectron peaks of O1s, O(KL1), Si2s, and Si2p Auger signals were identified. While the EDS spectrum can detect elements to a depth of micron, XPS only can detect elements to a depth of about 10 nm, which is less than the shell thickness of about 13 nm. In other words, XPS spectrum can detect the surface or shell elements of shell–core nanowires. Therefore, combining with the above analysis, the surface compound of shell–core nanowire can be supposed to be amorphous SiO2.

XPS scan spectrum of the as-synthesized sample sintered at 1500 °C
Figure 6 shows the TEM image and the EDS spectrum of the as-synthesized sample sintered at the initial reaction stage (irradiated by laser beam with the power density of 5.9 ± 0.1 W/mm2). It is found that some nanowires grew up from the nanospheres. From the EDS spectrum of the nanospheres, it can be inferred that the main elements were Si along with a little O and C content. It indicated that Si phase was formed at the initial reaction stage irradiated by high power density.

a TEM image of the sample irradiated at 5.9 ± 0.1 W/mm2 and b EDS spectrum of spectrum 2
Figure 7 shows the mass change of SiO2@SiC nanowire and SiC powders after isothermal oxidation test at different temperatures. It is demonstrated that SiO2@SiC nanowire had no obvious mass change after isothermal oxidation test except for about 4.5% mass loss at 1600 °C. However, pure SiC powders had large mass change after isothermal oxidation test at different temperatures. There was an increase of 24.7% in mass for pure SiC at 1400 °C, while the mass change decreased to only 13.4% at 1600 °C.

Mass change curves of the samples after isothermal oxidation test at different temperatures
Figure 8 demonstrates the room-temperature PL spectrum of the as-synthesized SiO2@SiC nanowire under 260-nm ultraviolet fluorescent light excitation. It shows an UV emission peak at 320–400 nm centered around 365 nm (3.40 eV) and the other at 420–500 nm centered around 453 nm (2.74 eV). Compared with the previously reported photoluminescence from the β-SiC monocrystal (2.39 eV, 518 nm) [23], the emission peaks were all obviously blue-shifted. Although the emission peak centered around 453 nm (2.74 eV) was slightly red-shifted, the UV emission peak centered around 365 nm (3.40 eV) was intensively blue-shifted compared with the SiC/SiO2 core–shell (2.82 eV, 440 nm) fabricated by molten salt-mediated carbothermal reduction method [24].

Room-temperature PL spectrum of the as-synthesized SiO2@SiC nanowires
Discussion
From the FESEM images and the EDS patterns of the as-synthesized samples sintered by laser sintering at 1300 and 1500 °C, it can be concluded that the sintering temperature has significant effects on the morphology and composition of the samples. Combining the XRD, EDS, and XPS analyses, SiC powders sintered by laser sintering at 1300 °C are supposed to be oxidized into SiO2 by the following reaction equation:
In this condition, a part of SiC particles were converted into amorphous SiO2. Thus, only some SiC crystal phase was detected in the XRD pattern of the as-synthesized samples sintered at 1300 °C.
When the sintering temperature was increased to 1500 °C, SiO2@SiC shell–core nanowires were formed. For the growth mechanism of one-dimensional nanostructures prepared by thermal evaporation method demonstrated previously, gaseous phase was essential and played an important role in the formation process. In this study, although the growth condition was different and no catalyst was employed, the gaseous phases involved could be generated from the reaction of the raw materials. The growth mechanism of the as-synthesized SiO2@SiC shell–core nanowires was schematically illustrated in Fig. 9 and discussed in the following sections.

Schematic illustration of the growth mode of SiO2@SiC nanowires
It is worth noting that Si phase was detected in the as-synthesized samples sintered at 1500 °C. As we all know, the melting point of Si is only 1410 °C. That is to say, liquid-phase Si was in situ formed at that moment, which was supposed to be generated by the reaction in Eq. 2:
Simultaneously, SiC powders were easily oxidized into gaseous SiO and CO by laser sintering at this high temperature, which can be expressed using the reaction in Eq. 3:
The Si droplets itself could partly be oxidized into SiO vapor. Thus, the Si droplets could provide abundant active sites for the absorption of gaseous SiO and CO, which acted as nucleation sites (Fig. 9II, III). Then liquid Si–O–C droplets were formed owing to the dissolution of gaseous SiO and CO as they reached the gas–liquid interface. The generated SiO and CO reacted with each other to regenerate SiC nanowires by reaction (4):
Hence, the nucleation of SiC would start, followed by the formation of a short nanowire (Fig. 9IV, V).
After SiC nanowires grew out of the Si–O–C droplets, SiO and CO continuously reacted to form SiC that deposit on the tip of SiC nanowires, which could promote the growth of SiC nanowires with a large length–diameter ratio. During the cooling stage, amorphous SiO2 could be formed on the surface of SiC nanowires by reaction (5):
The amorphous SiO2 shell was eventually formed on the surface of SiC core, which results in the formation of SiO2@SiC shell–core nanowires (Fig. 9VI). Therefore, the growth process of SiC nanowires was controlled by a combination of SLS (solid–liquid–solid) and VLS (vapor–liquid–solid) in situ growth mechanisms, which might be combined into SLVS (solid–liquid–vapor–solid) in situ growth mechanism. However, the liquid phase was not the catalyst droplets as used in conventional VLS growth mechanism, but the Si droplets that formed in situ from SiC raw materials.
Based on the above analyses, it was concluded that SiO2@SiC shell–core nanowires could be obtained without using any catalyst only by controlling the nucleation probability. A previous theory has demonstrated the possibility of nucleation on a whisker/wire surface, which can be expressed by the following equation [25]:
where P N is the nucleation probability, B is a constant, σ is the surface energy of whiskers, k is the Boltzmann constant, T is the absolute temperature, and α is the supersaturation ratio defined as α = P/P 0, where P is the actual vapor pressure and P 0 is the equilibrium vapor pressure corresponding to temperature T. According to Eq. (6), a higher temperature and a larger supersaturation ratio are beneficial to the increase of the nucleation probability of whiskers. Therefore, SiC nanowires were more easily formed at 1500 °C than 1300 °C.
The SiO2@SiC shell–core nanowires reveal good high-temperature oxidation resistance, as illustrated in Fig. 6. The 4.5% mass loss of the SiO2@SiC nanowires at 1600 °C could be attributed to the CO volatilization generated by the reaction in Eq. 7:
However, the mass of pure SiC increased by 24.7% at 1400 °C and then decreased to 13.4% at 1600 °C. The mass increase before 1400 °C was due to the formation of SiO2 by the oxidation of SiC, which can be expressed using Eq. 1. Above 1400 °C, the mass began to decrease. This indicated that the gas-generating reaction started from SiC in the reactions expressed using Eqs. 2 and 7. In a word, SiO2@SiC nanowires were much more thermodynamically stable than pure SiC. The main reason is the protective effect of inert SiO2 shell formed in situ on the surface of SiC nanowires.
The considerable blue-shifted emission peaks of the SiO2@SiC nanowires are shown in Fig. 7. There exist two obvious blue-shifted emission peaks centered around 365 nm (3.40 eV) and 453 nm (2.74 eV), respectively. So far, the PL mechanism of the SiO2@SiC shell–core structure is still not very clear because previous researches illustrated different PL mechanisms. The wavelength (band gap) of the emission peaks and the PL mechanism of the SiO2@SiC shell–core structure in different references are listed in Table 1.
Due to the remarkable nanostructure of SiO2@SiC shell–core nanowires used in this work, the emission peaks centered around 365 nm (3.40 eV) and 453 nm (2.74 eV) could be attributed to the oxygen vacancy of silicon long-pair centers such as O–Si–O and O–Si–C–O that are distributed at the SiO2@SiC interface boundary, the morphology, and structural defects such as stacking faults and twins, respectively. In conclusion, the prepared SiO2@SiC shell–core nanowires with preferable photoluminescence property and good thermodynamic stability are potential candidate materials for the fabrication of blue–violet emitting devices.
Conclusions
The obtained SiO2@SiC shell–core nanowires with a diameter of about 60 nm have been synthesized by laser sintering at about 1500 °C without any catalyst. Based on SLVS growth mechanism, the nucleation probability and the yield obtained increased as the sintering temperature increased. Compared with pure SiC, the SiO2@SiC nanowires were much more thermodynamically stable, which was attributed to the protective effect of inert SiO2 shell with a thickness of about 13 nm formed in situ on the surface of SiC nanowires. The SiO2@SiC nanowires showed two distinct emission peaks, i.e., the UV emission peak centered around 365 nm (3.40 eV) and another emission peak centered around 453 nm (2.74 eV), both of which were obviously blue-shifted compared with the previously reported photoluminescence from the β-SiC monocrystal (2.39 eV, 518 nm).
References
Ishikawa T, Kohtoku Y, Kumagawa K et al (1998) High-strength alkali-resistant sintered SiC fibre stable to 2200 °C. Nature 391:773–775
Herzog A, Thunemann M, Vogt U et al (2005) Novel application of ceramic precursors for the fabrication of composites. J Eur Ceram Soc 25:187–192
Wu RB, Zhou K, Yue CY et al (2015) Recent progress in synthesis, properties and potential application of SiC nanomaterials. Prog Mater Sci 72:1–60
Pourasad J, Ehsani N, Khalifesoltani SA (2016) Preparation and characterization of SiO2 thin film and SiC nanofibers to improve of graphite oxidation resistance. J Eur Ceram Soc 36:3947–3956
Meng A, Li ZJ, Zhang JL et al (2015) Synthesis and raman scattering of β-SiC/SiO2 core-shell nanowires. J Cryst Growth 308:263–268
Liu XM, Yao KF (2005) Large-scale synthesis and photoluminescence properties of SiC/SiOx nanocables. Nanotechnology 16:2932–2935
Li KZ, Weil J, Li HJ et al (2007) Photoluminescence of hexagonal-shaped SiC nanowires prepared by sol-gel process. Mater Sci Eng A 460:233–237
Seong HK, Choi HJ, Lee SK et al (2004) Optical and electrical transport properties in silicon carbide nanowires. Appl Phys Lett 85:1256–1258
Ye HH, Titchenal N, Gogotsi Y et al (2005) SiC nanowires synthesized from electrospun nanofiber templates. Adv Mater 17:1531–1535
Li ZJ, Gao WD, Meng AL et al (2009) Large-scale synthesis and raman and photoluminescence properties of single crystalline β-SiC nanowires periodically wrapped by amorphous SiO2 nanospheres 2. J Phys Chem C 113:91–96
Wang ZH, Dai Z, Gao RP et al (2000) Side-by-side silicon carbide-silica biaxial nanowires: synthesis, structure, and mechanical properties. Appl Phys Lett 77:3349–3351
Ryu Y, Tak Y, Yong K (2005) Direct growth of core-shell SiC-SiO2 nanowires and field emission characteristics. Nanotechnology 16:S370–S374
Zhang J, Yan S, Jia Q et al (2016) Preparation of SiC/SiO2 core-shell nanowires via molten salt mediated carbothermal reduction route. Phys E 80:19–24
Wang CS, Zhang JL, Meng AL et al (2007) Large-scale synthesis of β-SiC/SiOx coaxial nanocables by chemical vapor reaction approach. Phys E 39:128–132
Zhang HF, Wang CM, Wang LS (2002) Helical crystalline SiC/SiO2 core shell nanowires. Nano Lett 2:941–944
Chen K, Fang M, Huang Z et al (2013) Catalytic synthesis and growth mechanism of SiC@SiO2 nanowires and their photoluminescence properties. CrystEngComm 15:9032–9038
Li YB, Xie SS, Zou XP et al (2001) Large-scale synthesis of β-SiC nanorods in the arc-discharge. J Cryst Growth 223:125–128
Oliveira TC, Silva MS, Jesus LM et al (2014) Laser sintering and radioluminescence emission of pure and doped Y2O3 ceramics. Ceram Int 40:16209–16212
Macedo ZS, Silva RS, Valerio MEG (2004) Laser-sintered bismuth germanate ceramics as scintillator devices. J Am Ceram Soc 87:1076–1081
Sampaio DV, Souza NRS, Santos JCA et al (2014) Translucent and persistent luminescent SrAl2O4: Eu2+Dy3+ ceramics. Ceram Int 42:4306–4312
Jesus LM, Santos JCA, Sampaio DV et al (2016) Polymeric synthesis and conventional versus laser sintering of CaCu3Ti4O12 electroceramics: (micro)structures, phase development and dielectric properties. J Alloy Compd 654:482–490
Silva RS, Hernandes AC (2006) Laser sintering of BaTiO3 ceramics obtained from nanometric powders. Mater Sci Forum 514–516:1216–1220
Han W, Fan S, Li Q et al (1997) Continuous synthesis and characterization of silicon carbide nanorods. Chem Phys Lett 265:374–378
Zhang J, Jia Q, Zhang S et al (1997) One-step molten-salt-mediated preparation and luminescent properties of ultra-long SiC/SiO2 core-shell nanowires. Ceram Int 42:2227–2233
Dai ZR, Pan ZW, Wang ZL (2003) Novel nanostructures of functional oxides synthesized by thermal evaporation. Adv Funct Mater 13:9–24
Fabbri F, Rossi F, Attolini G et al (2012) Luminescence properties of SiC/SiO2 core–shell nanowires with different radial structure. Mater Lett 71:137–140
Kassiba A, Makowska-Janusik M, Boucle J (2002) Photoluminescence feature on the raman spectra of quasistoichiometric of SiC nanoparticles: experimental and numerical simulations. Phys Rev 66:155317
Nishikawa H, Shiryawa T, Hakamura R et al (1992) Photoluminescence from defect centers in high-purity silica glasses observed under 7.9-eV excitation. Phys Rev B 45:586–591
Yu DP, Hang QL, Ding Y et al (1998) Amorphous silica nanowires: intensive blue light emitters. Appl Phys Lett 73:3076–3078
Wei J, Li KZ, Li HJ et al (2008) Large-scale synthesis and photoluminescence properties of hexagonal-shaped SiC nanowires. J Alloy Compd 462:271–274
Acknowledgements
The authors acknowledge the financial support provided by the National Natural Science Foundation of China (Grant Nos. 51362022, 51302131) and the Natural Science Foundation of Jiangxi Province, China (Grant Nos. 20151BAB206015, 20161BAB206111).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, X., Huang, J., Lu, J. et al. Fabrication and characterization of SiO2@SiC shell–core nanowire prepared by laser sintering. J Mater Sci 52, 3344–3352 (2017). https://doi.org/10.1007/s10853-016-0622-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-0622-0