
Abstract
The wettability of ceramic and metallic solids by liquid metals is discussed with emphasis on the effect of interfacial reactions on the spreading kinetics and the final degree of wetting. Two types of reaction are considered, simple dissolution of the solid in the liquid and dissolution followed by formation of a new compound at the interface. The review includes results obtained during the last 15 years mainly by the Grenoble group.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
This review focuses on the wetting of high-temperature solids, mainly ceramics, by liquid metals and alloys. Many metal/ceramic couples are far from equilibrium and the resulting reactions can strongly modify the chemistry and the microstructure of the contact area. These changes affect both the spreading kinetics and the ultimate degree of wetting, and these aspects of reactive wetting can be exploited in practice to control wetting in different applications in materials science and processing: joining of solid components by brazing alloys [1, 2], processing of metal/ceramic composites by infiltration [3, 4] or selection of crucibles and moulds for molten metals (see for instance [5, 6]).
Data on the wetting of ceramic and metallic solids by liquid metals published until the end of the last century have been extensively reviewed in [7]. The present paper aims to review results obtained during the last 15 years, mainly, but not only, by the Grenoble group, especially results studying the relation between reactivity and wettability. Although this review focuses on liquid metal/ceramic systems, a limited number of results concerning liquid metal–solid metal systems are also given because they are useful in understanding bonding at metal/ceramic interfaces.
In the first section, after a brief overview of the fundamental equations of wetting and adhesion, the contact angles and spreading kinetics of non-reactive metals on different types of solids are briefly described. The second section concerns wetting in reactive systems where a layer of a new compound is formed at the interface. Two cases will be considered: in the first case, the compound is better wetted by a liquid than the initial substrate, while in the second case, the opposite situation occurs. The third section deals with dissolutive wetting, i.e. wetting accompanied by dissolution of the solid in the liquid.
The technique most widely used for wetting experiments at high temperatures is the sessile drop method. In its classical form, a small piece of solid sessile drop material, typically some tens or hundreds of mg, is placed on a substrate and then heated to the experimental temperature T above its melting temperature T m. Obviously, with this technique, the melting and wetting processes cannot be separated. This is possible using one of the following two variants. In the first (“dispensed drop” technique), a metal or alloy is melted in an unwetted and chemically inert ceramic crucible and is dispensed on the substrate surface through a thin capillary attached to the crucible by applying a back pressure of inert gas or using a piston. Another advantage of this technique is that oxide films on liquid metals are disrupted during dispensing. In the “transferred drop” technique, a sessile drop can be melted on an inert substrate, which is then raised so that the top surface of the drop contacts a fresh solid surface and subsequently lowered. Liquid is transferred to the top substrate provided this is better wetted than the donor. A detailed discussion on the different versions of the sessile drop technique is given in [8].
Most of the results presented below have been obtained by the “dispensed drop” version of the sessile drop technique.
Non-reactive wetting
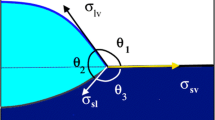
The intrinsic aptitude of a non-reactive liquid to wet a flat, smooth and chemically homogeneous solid surface is quantified by the value of Young’s contact angle θ Y, a unique characteristic of a solid S–liquid L–vapour V system (Fig. 1). θ Y enters into all model equations describing the wetting of liquids on real solid surfaces, i.e. surfaces with a certain roughness and degree of heterogeneity [7], as well as into equations modelling wetting in reactive liquid–solid systems (see the section “Reactive wetting”). θ Y is given by the classical equation of Young (Eq. 1) where the quantities σ SV and σ LV define the surface energy of the solid and liquid, respectively, and σ SL the solid/liquid interface energy.

Definition of the equilibrium contact angle θ. For a flat, smooth and chemically homogeneous solid surface, θ is Young’s contact angle θ Y
Moreover, by measuring θ Y and the surface energy of the liquid, Dupré’s adhesion energy of the system W a = σ SV + σ LV−σ SL can be evaluated through the Young–Dupré equation:
where the quantity W a characterizes the thermodynamic stability of interfaces between dissimilar materials.
Usual liquid metals are high-surface energy liquids. Their surface energy σ LV lies between 0.5 J m−2 for low melting point metals such as Pb and Sn and 2 J m−2 for high-melting point metals such as Fe and Mo [7]. These values, reflecting the high cohesion of metals due to their metallic (i.e., chemical) bonding, are one to two orders of magnitude greater than the surface energies of room temperature liquids in which bonding is achieved by weak, intermolecular interactions (i.e. physical interactions). Then, according to Eq. 2, good wetting (i.e. a contact angle of a few degrees or tens of degrees) of a liquid metal on a solid substrate can be observed if the adhesion energy is close to the cohesion energy of the liquid equal to 2σ LV. This is possible only if the interfacial bond is strong, i.e. chemical in nature. This condition is fulfilled for liquid metals on solid metals regardless of the miscibility between the liquid and the solid, because in this type of system, the interfacial bond is metallic. For instance, good wetting is observed for liquid Cu on solid Mo despite the absence of any miscibility in this system (Table 1). Liquid metals also wet semiconductors such as Ge or SiC because these solids, that are covalent in the bulk, are metallic in nature near the surface. Finally, liquid metals also wet ceramics such as carbides, nitrides or borides of transition metals because a significant part of the cohesion of these materials is provided by metallic bonds (see, for instance, Ref. [9]). Among the solids that are not wetted by non-reactive liquid metals are the different forms of carbon, the ionocovalent oxides and the predominantly covalent ceramics with a high band gap like BN. In these non-wetting systems, adhesion is provided by weak, van der Waals interactions.
Between systems with θ Y ≫ 90°, corresponding to weak, physical solid–liquid interactions, and systems with θ Y ≪ 90° (strong, chemical interactions), there are some liquid metal/solid oxide combinations where θ Y is in the range 80°–100°. These values correspond to an adhesion energy W a that is 2–2.5 times higher than the adhesion energy of non-reactive metals on oxides for the same σ LV value and reflect the development of moderate interactions of a chemical nature through the interface. Such interactions can result from two effects (see for a detailed discussion Ref. [10]): (i) chemisorption at the metal/oxide interface of oxygen dissolved in the bulk metal (a typical example is the (Ag + O)/Al2O3 system) and (ii) formation at the oxide side of the metal/oxide interface of a two-dimensional layer with a lower O/metal ratio than in the bulk oxide (typical example being the Al/Al2O3 system).
The spreading of liquid metals and alloys in non-reactive systems is a very fast process because, as the viscosity of this type of liquid is very low, a few mPas−1 [11], the viscous friction in the droplet during spreading is negligible [7, 12]. Thus for couples with θ Y not too low (say θ Y > 20°), the “spreading time” t spr (defined as the time needed for a millimetre-sized droplet to attain the equilibrium contact angle) is around 10 ms [13–15] (see an example in Fig. 16). Significantly higher spreading times can be observed in systems with equilibrium contact angles close to zero. In this case, instead of a drop, the liquid rapidly forms a film (Fig. 2) in which the viscous friction during spreading is no longer negligible [16].

Spreading of a CuAg alloy saturated in Cu on monocrystalline Cu studied by the dispensed drop technique. The diameter of the droplet before wetting is 0.9 mm. Reproduced from [46]
Reactive wetting with formation of a new compound at the interface
Experimental facts
An example of wetting for this type of reactivity is depicted in Fig. 3 for an Ag-1at%Zr alloy on aluminium nitride. While with pure silver, the contact angle is 137° and does not change with holding time, in the case of the alloy, the initial non-wetting contact angle decreases dramatically and reaches a steady contact angle close to 25° in a few 100 s. In this example, the reaction, leading to formation at the interface of a ZrN layer, has two main effects which are typical in reactive alloy/ceramic systems: (i) It produces a considerable improvement in wetting corresponding to a high increase in the adhesion energy (in this case by a factor 7) and to a transition from thermodynamically weak to thermodynamically strong interface. This effect can be explained by the bonding characteristics of ZrN which is a partly metallic ceramic. (ii) It leads to a substantial increase in the spreading time from a few ms for the non-reactive liquid to hundreds of seconds for the reactive one.

Spreading kinetics of Ag-1at%Zr alloys on AlN at 970 °C by the sessile drop technique (left), SEM image of a cross section of Ag-1%Zr on AlN (right, top) and qualitative compositional line analysis (right, bottom). Adapted from [47]
Modelling
Two different approaches have been proposed in order to explain this type of reactive wetting. In the first (the reaction product control (RPC) model), both the final degree of wetting and the spreading rate are determined by the reaction leading to the formation of the new compound at the triple line [7, 17]. In the second approach, published in 1998, the driving force for the reactive wetting is assumed to be caused mainly by adsorption, whereas spreading kinetics should be controlled by the migration of a ridge formed at the solid–liquid–vapour triple line [18]. However, detailed comparisons between predictions made by this model and experimental data have not confirmed this approach [17, 19].
According to the RPC model, the initial contact angle θ 0, formed in a few ms, is the contact angle on the surface of the unreacted ceramic substrate S (θ 0 = θ S) (Fig. 4a). After a transient stage, a quasi-state configuration is established at the triple line where the advance of the liquid is hindered by the presence of a non-wettable substrate in front of the triple line (Fig. 4b). Thus, the only way to move ahead is by lateral growth of the wettable reaction product layer P until the macroscopic contact angle equals the equilibrium contact angle of the liquid on the reaction product (θ F = θ P) (Fig. 4c). The example given in Fig. 4 (bottom) is for a CuSi alloy on vitreous carbon forming a silicon carbide interfacial layer. Note that the RPC model supposes that the thickness of the reaction product layer is small in comparison with droplet size. This is a reasonable assumption for most metal/ceramic systems where when the capillary equilibrium is attained this thickness is a few microns, i.e. three orders of magnitude lower than the size of droplets used in wettability experiments.

Top schematic representation of the “Reaction Product Control” model. Initial contact angle θ 0 (a); quasi-state configuration (b) and final contact angle θ F (c). Bottom contact angle θ versus logt for a Cu-40at%Si alloy on vitreous carbon at 1180 °C (P = SiC). Dispensed drop technique. The arrow shows the instant of detachment of the droplet from the capillary. Reproduced from [26]
The spreading time t spr = t(θ = θ F ) in reactive metal/ceramic systems is in the range 10–104 s, i.e. several orders of magnitude higher than t spr ≈ 10−2 s observed for non-reactive metals. Therefore, in a given reactive system, the spreading rate of the reactive stage is limited by the interfacial reaction itself, especially by the interfacial reaction at the triple line where the liquid can continuously contact a fresh substrate surface. The rate of the interfacial reaction at the triple line, in turn, may be controlled by the slower of two successive phenomena that intervene in the reaction process: diffusive transport of reacting species from the liquid bulk to the triple line and local reaction kinetics at the triple line.
Diffusion controlled wetting was modelled in 1997 by Mortensen et al. who established an analytical expression giving the spreading rate U = dR/dt (R being the drop base radius) as a function of the instantaneous contact angle θ [20]. The validity of the model was discussed using results obtained for Cu–Cr [21] and Cu–Sn–Ti [22] alloys on vitreous carbon, where the concentration of the reactive alloying element (Cr and Ti, respectively) was in the range of a few per cent. An improvement of the model was published in [23] where the authors used a numerical approach in order to take into account the effect on the spreading rate of the interfacial reaction occurring behind the triple line.
In the case of control by the local reaction at the triple line, once a quasi-steady configuration is established at the triple line, the rate of reaction and hence the triple line velocity are expected to be constant with time. Since the first experimental evidence observed for aluminium on carbon [24], nearly constant triple line velocities U have been observed for different systems like CuSi alloys on oxidized SiC [25], in the CuSi/C [26] and NiSi/C [19] systems, for Ti-containing Ni-based alloys on AlN [27] and for Si on Si3N4 [28]. An example is given in Fig. 5 for a silicon carbide forming AlSi alloy on vitreous carbon. After a transient state, where the contact angle decreases rapidly from 160° to 90°, the spreading rate U continues to decrease but only slightly (“quasi-linear spreading”) and tends towards a constant value when θ tends towards θ N. As the micrographs of Fig. 6 show, the microstructure of the reaction product (SiC) is equiaxial at the centre of the interface and columnar in the “quasi-linear wetting” stage where the SiC dendrite growth direction is close to the direction of movement of the triple line. Note that at t > t N, the spreading rate is very small (0.1 μm/s) but not nil. This final spreading stage can be more easily distinguished in Fig. 7. As argued in [29], at t < t N direct contact between the liquid and initial substrate leads to strong coupling between the local chemical reaction and spreading kinetics. Conversely, at t > t N direct contact no longer exists, coupling is weak and as a consequence the spreading rate is small (for more details on the stage at t > t N see Ref [29]).

Contact angle θ, drop base diameter d and spreading rate U versus time for an experiment conducted by the dispensed drop technique on vitreous carbon with an Al-25at%Si alloy at 1000 °C. Reproduced from [30]

Top view, after removal of the AlSi alloy, of the reaction layers close to the triple line for experiments interrupted after 60 and 600 s of spreading (a, b, respectively). TL triple line. T = 1000 °C, vitreous carbon. Reproduced from [30]

Napierian logarithm of cosθ F−cosθ versus time t for the Ni-63at%Si alloy/Cv system at different temperatures studied by the dispensed drop technique. The black arrows show the time t N at which the sharp change in the slope of R versus t curves occurs, indicating the transition from strong to weak coupling between the chemical reaction and the wetting process at TL. The perturbations at around 50 s are due to droplet detachment from the capillary. Reproduced from [48]
In [26], a model, leading to analytical expressions of contact angle as a function of time, has been developed for the quasi-linear wetting stage assuming that the reaction rate at the triple line parallel to the interface is controlled by the dissolution process, i.e. by the process of atom transfer occurring at the substrate/alloy interface.
The following equation for the change in contact angle with time t was derived:
where k is proportional to the kinetic constant k d and to the driving force Δμ of the dissolution process at the TL. For silicon-containing alloys in contact with carbon substrates Δμ is the difference between the chemical potential of carbon in the solid and in the liquid and is related to the activity of Si in the alloy according to Eq. 4.
The subscript I indicates the minimum value of reactive element Si for the formation of the reaction product SiC.
According to Eq. 3, logarithmic plots of \( \cos \theta_{\text{F}} - \cos \theta \) versus time would be linear with a slope equal to minus k. An example is given in Fig. 7 for Ni-63at%Si alloy on vitreous carbon at different temperatures. The experimental data agree with the linearity predictions in a large domain of theta. The sharp change in the slope of straight lines reflects the high value of the activation energy of the dissolution process (about 300 kJ mol−1 in this case). The kinetic constant k d of the dissolution reaction of the substrate is also highly sensitive to the crystallographic structure of the initial substrate. This explains the wide variation in triple line velocities observed for silicon carbide forming AlSi alloys on different kinds of carbon: vitreous carbon, pyrocarbon, and pseudo-monocrystalline graphite [30]. Moreover, Eq. 4 predicts a significant but limited effect of alloy concentration on the spreading rate in the quasi-linear stage, in agreement with the experimental results [30].
Interfacial chemistry versus wetting
The relation between the interfacial chemistry and wetting was studied for CuAg alloys containing a few per cent of Ti on alumina substrates, a system presenting a high interest in brazing of alumina components [31]. The effect of Ti on wetting is illustrated by the sessile drop experiment depicted in Fig. 8 in which a CuAg-1.75wt%Ti alloy was processed in situ, by placing a small quantity of Ti on top of a piece of AgCu on an alumina substrate [32].

Contact angle as a function of time for two CuAgTi droplets of different composition processed in situ on monocrystalline alumina in He at 900 °C. Reproduced from [32]
The large, non-wetting, contact angle observed at melting, which is typical of a non-reactive noble metal on an oxide, is the contact angle of CuAg alloy on alumina. This angle remains constant for several minutes, the time needed for Ti to dissolve in the liquid and reach the metal/oxide interface by diffusion. Afterwards, the contact angle decreases rapidly towards a value close to 10°, which corresponds to excellent wetting.
The micrograph in Fig. 9 presents the interface formed between the alloy and alumina at the end of the wetting experiment. The reactive interface is composed of two layers. The layer formed in contact with the liquid alloy was identified as the Cu3Ti3O compound. The low final contact angle observed in this system is explained by the metallic character of this compound. A thin, 1- to 2-micron layer is also formed in contact with alumina and identified as the Ti2O suboxide. By decreasing the Ti concentration in the alloy to 0.42 wt%, only the Ti suboxide was formed at the interface, and, as a consequence, a very significant increase in the equilibrium contact angle was observed. Even higher contact angles can be observed with further reduction in Ti concentration or, more generally, of titanium activity in the alloy, which is in reality the relevant thermodynamic quantity involved in the interfacial reactivity. Indeed, contact angles in the range 90°–110° have been observed by Kritsalis et al. for NiPd–Ti alloys containing several per cent of Ti. In this system, the titanium activity is extremely low due to very strong Ni–Ti and Pd–Ti interactions in the alloy, thus leading to formation at the interface of Ti oxides with a high degree of oxidation such us Ti5O9 [33].

Cross section of a CuAg-3at%Ti/Al2O3 interface, after 30 min at 900 °C in He. Reproduced from [32]
In experiments depicted in Fig. 8, melting and spreading occur simultaneously. Therefore, these experiments do not provide proper information on spreading kinetics in this system. This information was obtained by Kozlova et al. [34] thanks to experiments performed by the dispensed drop technique. An example of spreading curves is given in Fig. 10. As shown in Table 2, spreading in this system occurs in three successive stages. After initial, very fast non-reactive spreading (not shown in Fig. 10), the low final contact angles observed in the CuAgTi/alumina system are attained by two reactive stages. (i) The first reactive stage (until t*) is rapid and can be explained by the formation at the interface of Ti suboxide. The spreading rate in this stage is in agreement with a model of diffusion-limited spreading. (ii) The second reactive stage is much slower, driven by the formation of the Cu3Ti3O compound and controlled by the local chemical process at the triple line.

Contact angle and drop base diameter versus time for a CuAg-0.76wt%Ti alloy at 850 °C obtained by the dispensed drop technique (a) The same results presented using for time plotted on a logarithmic scale (b) The perturbation on the curves shown by an arrow corresponds to the moment of detachment of the drop from the capillary. Reproduced from [34]
When alumina (or another ceramic) is brazed to a metallic partner Me, the interactions between Me and the brazing alloy can affect the joint microstructure and the interfacial chemistry. Indeed, as the diffusion time in a liquid gap of 100 microns is only a few seconds, Me dissolves in the liquid braze and attains rapidly its equilibrium concentration. For instance, when one of the alumina pieces is replaced by a copper–nickel plate (Fig. 11), the dissolution of Ni and Cu from the plate in the liquid braze leads to a dramatic change in joint microstructure and also a net change in reactivity at the alumina/braze interface consisting of a decrease in the thickness of the interfacial reaction layer by one order of magnitude (from ~2 μm to ~200 nm). Moreover, instead of the wettable Cu3Ti3O compound formed at the interfaces in the alumina to alumina joint, high oxidation-level Ti oxides are formed in the case of CuNi/alumina joint, resulting in a very significant increase in contact angle towards 90° and ultimately in the mechanical weakening of the interface and of the actual joint [35]. From these results, it appears that when ceramics are brazed to metallic parts, allowance must be made not just for the well-known problem of thermo-mechanical compatibility between these two types of solid but also for the interactions between the metallic part and the braze that can affect wetting as well as the joint composition and microstructure.

Cross sections of alumina to alumina (a) and alumina to CuNi (b) plates. Assemblies brazed by a CuAg–Ti alloy (900 °C, 15 min). e denotes here the thickness of the interfacial reaction product. Reproduced from [35]
Reaction product less wetted than the initial substrate
In the previous examples, the reaction product is better wetted than the initial substrate. The RPC model also holds true when the opposite situation occurs, for instance, in the couples Ag/SiC, Cu/SiC and Au/TiC.
When a small piece of a metal such as Cu or Ni is placed on silicon carbide, a strong reaction between the metal and silicon carbide takes place [36, 37]. The reaction consists of SiC dissolution in the metal, while the excess carbon forms graphite precipitates close to the interface:
where the parenthesis means silicon dissolved in the metal. While liquid metals wet clean SiC surfaces, they do not wet graphite. Therefore, this reaction inhibits wetting (Fig. 12) and additionally leads to the formation of brittle graphite particles that are detrimental to mechanical properties. To avoid these effects, metal-silicon alloys are used instead of pure metal. For a given system, the silicon concentration is chosen such that the formation of graphite precipitates is suppressed. In this way, contact angles as low as 20°–40° were observed as well as mechanically strong metal/SiC interfaces [36]. These findings formed the basis of the development of new, high-temperature alloys for joining SiC components by brazing.

Left contact angle as a function of time and temperature for pure Cu on sintered SiC. The increase in temperature produces an increase in the size of the reactive zone (right) but does not change the contact angle. Right schematic cross section of the pure Cu-sintered SiC experiment showing the reaction zone. Reproduced from [37]
Pure Au does not wet TiC, having a contact angle of about 130° (Fig. 13), a value that is similar to that obtained for Au on carbon substrates. Because of the strong interaction between Au and Ti, a slight dissolution of Ti from the substrate to the liquid occurs. However, given that the solubility of C is much smaller than that of Ti, graphite precipitates at the surface and thus the measured contact angle of 130° is the contact angle of Au on graphite. Since the solubility of C in molten Ni and Fe (but not in Cu) is several orders of magnitude higher than in Au, small additions of these elements in Au increase the solubility of C significantly and, thereby, prevent the formation of the graphite interface layer on the TiC substrate. As a result, the experimental results show a dramatic decrease in contact angle with a few at% of Ni or Fe without any reaction product at the interface [38]. In this system, the action of Ni and Fe is to remove, by dissolution, the wetting barrier that inhibits spreading.

Contact angle between TiC and Au alloys at 1150 °C. Adapted from [38]
Metallic solids
As discussed in the section “Non-reactive wetting”, liquid metals wet metallic solids well even in the absence of any reactivity between the liquid and the solid (Table 1). For this reason, reactions leading to the formation of intermetallic compounds at the interface produce a significant but rather limited improvement in wetting [7]. However, at low temperatures, normal metallic solids are covered by thin oxide films a few nanometres thick, formed rapidly in contact with air at room temperature. Such oxides, that may be stable even in high vacuum or in low oxygen partial pressure gas mixtures, are not wetted by liquid metals. In this case, wetting can be significantly improved by interfacial reactions leading to the in situ replacement of the contaminated surface by a clean surface of an intermetallic compound. An example is given in [39]. At 400 °C molten lead wets well (θ ≈ 45°) clean, non-oxidized surfaces of Fe produced in situ by a prior heat treatment at 850 °C. However, when molten lead contacts Fe substrates heated directly to 400 °C, the contact angle is close to 90° and does not change with time, thus indicating that the oxide film on Fe is stable at this low temperature (Fig. 14 ). The initial contact angle formed by a drop of Sn on an oxidized Fe surface is close to 90°, as for Pb. However, in the case of Sn, due to the formation at the interface of the FeSn2 intermetallic compound, the contact angle starts to decrease and tends towards a value close to 30° in 1000 s. Hence, due to the disruption of oxide layers caused by the reaction, the final degree of wetting in liquid metal/metallic solid systems with significant reactivity is generally much less sensitive to environmental factors than in non-reactive systems.

Left change with time of contact angles of pure Sn and Pb under gettered He at 400 °C. Direct heating at 400 °C (dispensed drop technique). Right schematic description of the region close to the triple line for an oxidized metal: a in the case of a non-reactive liquid metal B-solid metal A couple and b in the case of formation of a layer of intermetallic compound AB n at the interface. The isolated particles come from breaking up of the oxide layer. Reproduced from [39]
Dissolutive wetting
Mechanism
Extensive dissolution of a solid in a liquid is a phenomenon occurring in many liquid metal/solid metal systems as well as in some metal/ceramic ones, such as Ni/C [7], Ni/HfB2 [40] and Ni/SiC [36]. Dissolution can reduce the observed (or visible) contact angle through two effects. First, by decreasing, the surface tension of the liquid which occurs when the surface tension of a component of the solid is much lower than that of the liquid. Second, by forming under the droplet, a crater such that the observed contact angle θ ap is lower than the true angle θ F formed at the solid/liquid/vapour junction (Fig. 15).

Dissolutive wetting: Wetting of a liquid metal B leads rapidly, within a few ms, to a first contact angle θ 0 corresponding to the intrinsic contact angle of pure B in metastable equilibrium with A. Then the wetting process continues with dissolution of A in B, leading to formation of a crater until saturation of B in A. Reproduced from [15]
Protsenko et al. [15] proposed that the formation of a crater results in further wetting of the liquid on the substrate surface in order to maintain the capillary equilibrium at the three-phase junction. Accordingly, θ F should be the intrinsic contact angle of a B liquid saturated in A on solid A measured on an inclined solid surface. Conversely, other authors consider that this angle does not result from the equilibrium of surface energies but by kinetic factors related to diffusion of dissolved species from the interface to the liquid bulk [41, 42]. As for the spreading time, experimental values of this quantity are much higher than for non-reactive spreading [15, 42]. An example is given in Fig. 16 for pure Cu on solid Si (t spr ≈ 1 s) compared to copper presaturated in Si (t spr ≈ 10−2s). This fact strongly suggests that the spreading rate in dissolutive wetting is controlled by the transport of dissolving species in the bulk liquid.

Visible contact angle (left) and drop base diameter (right) as a function of time plotted on a logarithmic scale for Cu and Cu presaturated in Si on a (001) Si surface at 1100 °C. Dispensed drop technique. Reproduced from [15]
On this basis, a simple equation was established in [15] according to which the dissolutive spreading rate dR/dt varies proportionally to the difference between the instantaneous and final visible contact angle. When the transport of dissolving species is assumed to occur by Si diffusion, the experimental data for the liquid Cu/solid Si system fit this equation well but with an apparent diffusion coefficient much higher than the typical values of diffusion coefficient in liquid alloys. It is argued that Marangoni convection, due to composition gradients along the drop upper surface, is responsible for the high value of the apparent diffusion coefficient obtained by fitting.
The role of dissolution in the wettability of composite surfaces
A new effect of dissolution on wettability was recently highlighted by Lai et al. [43] in wetting experiments of two-phase composite Cu–Fe substrates by molten Sn.
When a solid α contains a dispersion of particles β, the equilibrium contact angle θ C on the heterogeneous surface is given by Cassie’s equation:
where f α is the surface area fraction of the matrix. However, in [43], it was found that the curve of contact angle versus the surface fraction of composite components passes through a minimum behaviour that cannot be interpreted by Eq. 6. It was shown that the enhanced wetting observed on the composite substrate (Fig. 17) can be explained by the dissolution contrast of Cu and Fe phases, leading to increasing interfacial roughness, thus providing an additional driving force for wetting.

Top view of areas wetted by Sn on Cu, θ = 25°(a), 50Cu–50Fe, θ = 5°(b) and Fe θ = 47°(c). Reproduced from [43]
Dissolution versus formation of a new compound
An example which illustrates the two types of reactive wetting is given by results obtained by the dispensed drop technique with an Au-40at%Ni alloy on ZrB2 [44]. The experiments were performed at two temperatures, 980° and 1170 °C. A nickel boride is formed at the alloy/ZrB2 interface at 980 °C, while only dissolution of ZrB2 into the liquid alloy occurs at 1170 °C. Thus, by varying the temperature, it is possible to change the type of metal-ceramic reactivity by keeping the composition of the metallic phase constant.
Figure 18 gives the time-dependent change of drop base diameter in an experiment at 1170 °C, as well as during the subsequent cooling. Due to the metal-like character of ZrB2, the initial contact angle is low (28°) and decreases slightly to 25° thanks to substrate dissolution. Further spreading was observed when the temperature decreased to values where formation of the new compound (Ni2B) becomes possible.

Au-40at%Ni/ZrB2 system. Top drop base radius and temperature as a function of time. The numbers on the drop base radius curve represent the corresponding values of the contact angle. Bottom micrograph of the interface close to the triple line formed during cooling. 1 and 2 indicate the positions of the triple line before and after cooling. The crater on the left of the arrow 1 is due to dissolution. Reproduced from [44]
The micrograph of the region near the triple line (Fig. 18) shows the position of the triple line before cooling, separating the dissolution cavity formed at 1170 °C from the interface zone produced at lower temperature. The reaction product layer covering the interface, a few microns thick, can also be distinguished. From these observations, it is easy to understand the unusual result depicted in Fig. 18, namely that a decrease in temperature improves wettability.
In the previous example, the formation of a new compound at the interface leads to a net, but limited, improvement in wetting. This is because both the initial solid (ZrB2) and the reaction product (nickel boride) are metallic in character and thus wettable by liquid metals. A much more pronounced improvement is observed in the example depicted in Fig. 19 for two Ni–Si alloys on graphite at 1270 °C [45]. For the Ni-21at%Si alloy, only a limited dissolution of graphite in the liquid takes place. The steady contact angle attained a few minutes after melting is close to 120°. Completely different behaviour is exhibited by a Ni-47at%Si alloy, leading to the formation of wettable SiC at the interface. The alloy was shown to wet the graphite with a steady contact angle of 40°. This good wetting leads to infiltration of porous graphite by the liquid alloy.

Micrographs of two NiSi alloy/graphite sectioned samples. Left Ni-21at%Si, general view (a), alloy/carbon interface (b). Only a limited dissolution of graphite is observed. Right Ni-47at%Si, general view (a). An area of the infiltrated zone (b). All graphite particles are covered by a thin, submicronic layer of SiC. Reproduced from [45]
Conclusions and perspectives
During the last 15 years, a large number of studies have been published on the fundamentals of reactive wetting. Some have focused on the dissolutive wetting, studied using simple model systems. Although these studies have contributed significantly to improving our knowledge of dissolutive wetting, more work is needed to clarify points where diverging opinions persist, especially on the driving force of this type of wetting. As for the spreading kinetics, Marangoni convection seems to play a key role in mass transport. Further progress on this subject requires numerical simulations of the spreading process.
For reactive systems where wetting is accompanied by the formation of a new compound at the interface, a first version of the RPC model was proposed in [24]. The model was greatly improved thanks to the analytical approach proposed in [26] for the case where spreading kinetics is limited by the local chemical process at the triple line. This approach completes the previously published similar study of Mortensen et al. [20] for diffusion controlled reactive spreading.
The RPC model has been applied with success in the analysis of experimental data obtained with various ceramic substrates, namely the different types of carbon, oxides, nitrides or borides. A main assumption in the model is that the new compound growth process takes place inside a zone of submicronic size localized around the solid–liquid–vapour junction, where the reactive element contained in the liquid has direct access to the solid substrate. This seems to be a reasonable assumption when experiments are performed in neutral gas or in vacuum but at low or moderate temperatures. However, experiments performed with the Si/graphite couple in high vacuum at 1430 °C showed different wetting kinetics compared to that observed in neutral gas [5]. In high vacuum, the transport of a reactive solute through the vapour can modify the surface chemistry of the substrate in front of the triple line and thus enhance spreading. A similar effect is expected to occur under an inert gas but at higher temperatures. A general description of reactive wetting taking into account both the localised and delocalised reaction is still lacking.
As for the experimental methods used in wettability studies, great improvements are expected to be made in the very near future, including the development of new devices where sessile drop experiments can be coupled in the same chamber with high temperature surface characterization techniques (for instance by Auger, XPS spectroscopy or SEM). Performing surface characterization (either chemical or microstructural) at high temperatures is a difficult but very exciting objective.
References
Paponetti CA, Sapp M (2007) Basics of brazing. In: AWS C3 Committee on Brazing and Soldering (ed) AWC brazing handbook, s.l., vol 1. American Welding Society, New York, pp 1–20
Seculic D (ed) (2013) Advances in brazing: science, technology and applications. Woodhead Publishing, Oxford
Evans A, San Marchi C, Mortensen A (2003) Metal matrix composites in industry: an introduction and a survey. Kluwer Academic Publishers, Dordrecht
Einset EO (1996) Capillary interaction rates into porous media with applications to Silcomp processing. J Am Ceram Soc 79:333–338
Israel R, Voytovych R, Protsenko P, Drevet B, Camel D, Eustathopoulos N (2010) Capillary interactions between molten Si and porous graphite. J Mater Sci 45:2210–2217. doi:10.1007/s10853-009-3889-6
Huguet C, Deschamps C, Voytovych R, Drevet B, Camel D, Eustathopoulos N (2014) Initial stages of silicon-crusible interactions in crystallisation of solar grade silicon: kinetics of coating infiltration. Acta Mater 76:151–167
Eustathopoulos N, Drevet B, Nicholas M (1999) Wettability at high temperatures. Pergamon Materials Series, Oxford
Eustathopoulos N, Sobczak N, Passerone A, Nogi K (2005) Measurement of contact angle and work of adhesion at high temperature. J Mater Sci 40:2271–2280. doi:10.1007/s10853-005-1945-4
Passerone A, Muolo ML, Novakovic D, Passerone D (2007) Liquid metal/ceramic intercations in the (Cu, Ag, Au)/ZrB2 systems. J Eur Ceram Soc 27:3277–3285
Eustathopoulos N, Drevet B, Muolo ML (2001) The oxygen-wetting transition in metal/oxide systems. Mater Sci Eng A 300:34–40
Batezzati L, Greer LA (1989) The viscosity of liquid metals and alloys. Acta Metall 37:1791–1802
Kumar G, Prabhu KN (2007) Review of non-reactive and reactive wetting of liquids on surfaces. Adv Colloid Interface Sci 133:61–89
Saiz E, Tomsia AP (2004) Atomic dynamics and Marangoni films during liquid-metal spreading. Nat Mater 3:903–909
Saiz E, Tomsia AP, Rauch N, Scheu C, Ruehle M, Benhassine M, Seveno D, de Conick J, Lopez-Esteban S (2007) Non-reactive spreading at high temperature: molten metals and oxides on molybdenum. Phys Rev E 76:041602–041615
Protsenko P, Garandet JP, Voytovych R, Eustathopoulos N (2010) Thermodynamics and kinetics of dissolutive wetting of Si by liquid Cu. Acta Mater 28:6565–6574
De Gennes PG (1985) Wetting: static and dynamics. Rev Mod Phys 57:827–863
Eustathopoulos N (2005) Progress in understanding and modelling reactive wetting of metals on ceramics. Curr Opin Solid State Mater Sci 9:152–160
Saiz E, Tomsia AP, Cannon RM (1998) Ridging effect on wetting and spreading of liquids on solids. Acta Mater 46:2349–2361
Bougiouri V, Voytovych R, Dezellus O, Eustathopoulos N (2007) Wetting and reactivity in Ni-Si/C system: experiments versus model predictions. J Mater Sci 42:2016–2023. doi:10.1007/s10853-006-1483-8
Mortensen A, Drevet B, Eustathopoulos N (1997) Kinetics of diffusion-limited spreading of sessile drops in reactive wetting. Scr Mater 36:645–651
Voitovitch R, Mortensen A, Hodaj F, Eustathopoulos N (1999) Diffusion-limited reactive wetting: study of spreading kinetics of Cu-Cr alloys on carbon substrates. Acta Mater 47:1117–1128
Dezellus O, Hodaj F, Mortensen A, Eustathopoulos N (2001) Diffusion-limited reactive wetting: spreading of Cu-Sn-Ti alloys on vitreous carbon. Scr Mater 44:2543–2549
Hodaj F, Dezellus O, Barbier JN, Mortensen A, Eustathopoulos N (2007) Diffusion-limited reactive wetting: effect of interfacial reaction behind the advancing triple line. J Mater Sci 42:8071–8082. doi:10.1007/s10853-007-1915-0
Landry K, Eustathopoulos N (1996) Dynamics of wetting in reactive metal/ceramic systems: linear spreading. Acta Mater 44:3923–3932
Dezellus O, Hodaj F, Rado C, Barbier JN, Eustathopoulos N (2002) Spreading of Cu-Si alloys on oxidized SiC in vacuum: experimental results and modeling. Acta Mater 50:979–991
Dezellus O, Hodaj F, Eustathopoulos N (2003) Progress in modelling of chemical-reaction limited wetting. J Eur Ceram Soc 23:2797–2803
Koltsov A, Dumond M, Hodaj F, Eustathopoulos N (2006) Influence of Ti on wetting of AlN by Ni-base alloys. Mater Sci Eng A A415:171–176
Drevet B, Voytovych R, Israel R, Eustathopoulos N (2009) Wetting and adhesion of Si on Si3N4 and BN substrates. J Eur Ceram Soc 29:2363–2367
Dezellus O, Eustathopoulos N (2010) Fundamental issues of reactive wetting by liquid. J Mater Sci 45:4256–4264. doi:10.1007/s10853-009-4128-x
Calderon NR, Voytovych R, Narciso J, Eustathopoulos N (2010) Wetting dynamics versus interfacial reactivity of Al-Si alloys on carbon. J Mater Sci 45:2150–2156. doi:10.1007/s10853-009-3909-6
Kozlova O, Braccini M, Voytovych R, Eustathopoulos N, Martinetti P, Devismes MF (2010) Brazing copper to alumina using reactive CuAgTi alloys. Acta Mater 58:1252–1260
Voytovych R, Robaut F, Eustathopoulos N (2006) The relation between wetting and interfacial chemistry in the CuAgTi/alumina system. Acta Mater 54:2205–2214
Kritsalis P, Drevet B, Valignat N, Eustathopoulos N (1994) Wetting transitions in reactive metal/oxide systems. Scr Metall Mater 30:1127–1132
Kozlova O, Voytovych R, Eustathopoulos N (2011) Initial stages of wetting of alumina by reactive CuAgTi alloys. Scr Mater 65:13–16
Valette C, Devismes MF, Voytovych R, Eustathopoulos N (2005) Interfacial reactions in alumina/CuAgTi braze/CuNi system. Scr Mater 52:1–6
Rado C, Kalogeropoulou S, Eustathopoulos N (1999) Wetting and bonding of Ni-Si alloys on silicon carbide. Acta Mater 47:461–473
Rado C, Drevet B, Eustathopoulos N (2000) The role of compound formation in reactive wetting: the Cu/SiC system. Acta Mater 48:4483–4491
Frage N, Froumin N, Dariel MP (2002) Wetting of TiC by non-reactive liquid metals. Acta Mater 50:237–245
Protsenko P, Terlain A, Traskine V, Eustathopoulos N (2001) The role of the intermetallics in metallic systems. Scr Mater 45:1439–1445
Passerone A, Muolo ML, Valenza F, Monteverde F, Sobczak N (2008) Wetting and interfacial phenomena in Ni-HfB2 systems. Acta Mater 57:356–364
Warren JA, Boettinger WJ, Roosen AR (1998) Modelling reactive wetting. Acta Mater 46:3247–3264
Yin L, Murray BT, Singler TJ (2006) Dissolutive wetting in the Bi-Sn system. Acta Mater 54:3561–3574
Lai QQ, Zhang L, Eustathopoulos N (2013) Enhanced wetting of dual-phase metallic solids by liquid metals: a new effect of interfacial reaction. Acta Mater 61:4127–4134
Voytovych R, Koltsov A, Hodaj F, Eustathopoulos N (2007) Reactive vs non-reactive wetting of ZrB2 by azeotropic Au-Ni. Acta Mater 55:6316–6321
Bougiouri V, Voytovych R, Rojo-Calderon N, Narciso J, Eustathopoulos N (2006) The role of the chemical reaction in the infiltration of porous carbon by NiSi alloys. Scr Mater 56:1875–1878
Kozlova O, Voytovych R, Protsenko P, Eustathopoulos N (2010) Non-reactive versus dissolutive wetting of Ag-Cu alloys on Cu substrates. J Mater Sci 28:2099–2105. doi:10.1007/s10853-009-3924-7
Koltsov A, Hodaj F, Eustathopoulos N, Dezellus A, Plaindoux P (2003) Wetting and interfacial reactivity in Ag-Zr/sintered AlN system. Scr Mater 48:351–357
Dezellus O, Hodaj F, Eustathopoulos N (2002) Chemical reaction limited spreading: the triple line velocity versus contact angle relation. Acta Mater 50:4741–4753
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Eustathopoulos, N., Voytovych, R. The role of reactivity in wetting by liquid metals: a review. J Mater Sci 51, 425–437 (2016). https://doi.org/10.1007/s10853-015-9331-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-015-9331-3