
Abstract
Glassy carbon nanofibers (g-CNFs) with diameter of ca. 45 nm were prepared from electrospun cellulose nanofibers (CelluNFs) by two sequential steps: stabilization and carbonization. The CelluNFs were stabilized at 400 °C and carbonized at 850 and 1400 °C to produce g-CNFs. The morphology and crystallographic structure of the precursor, stabilized and carbonized nanofibers were characterized by field emission scanning electron microscopy, transmission electron microscopy (TEM), selected-area electron diffraction, and Raman spectroscopy. It was found that the stabilization treatment was very effective in maintaining the nanofiber morphology of the final product g-CNFs. TEM images revealed that the g-CNFs carbonized at 1400 °C showed ribbon-like curved single carbon layer networks throughout the sample. These layers were randomly stacked without any particular sequence, and were looped. Raman spectra confirmed that the g-CNFs contained low content of graphitic crystallites. The I D/I G values indicated that most carbons of g-CNFs are in the amorphous state. The electrical conductivity was dependent on the graphitic microstructure in the g-CNF. It reached to 93.5 S/cm for the g-CNF carbonized at 1400 °C. The g-CNF with good conductivity may find applications in the carbon-based nanodevices.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Carbon nanostructures such as carbon nanofibers (CNFs) and carbon nanotubes (CNTs) have attracted great academic and industrial attention due to their unique anisotropic properties and a wide range of potential applications. Compared with other nanoscale carbon materials, CNFs are good candidate carbonaceous materials for filtration, catalytic support [1], electronic component [2], hydrogen storage [3], and biomedical applications [4], owing to their good conductivity, high mechanical strength, high aspect ratio, and large specific surface area. Before polyacrylonitrile (PAN) and pitch became dominant precursors of carbon fibers, cellulose fibers (cotton fibers, rayon filaments, and bamboo fibers) had been the main precursors of carbon fibers, which were then used as the filament of incandescent light [5, 6].
Because of the high and continuous increasing cost of PAN-based carbon fiber due to the fluctuations of the crude oil prices, and large demand of carbon fiber in the automobile, aircraft, sports, and military industries, a resurging interest in renewable cellulosic precursors is building up [6, 7]. Carbon fibers were prepared from the pyrolysis and graphitization of liquid crystalline cellulose fiber precursor in the temperature range of 400–2500 °C. A graphitic structure was developed after the fibers were thermally treated at 2500 °C. They showed skin–core structure, and had moduli of ~140 and 40 GPa for the skin and the core, respectively [8]. The same group fabricated CNFs from electrospun cellulose nanofibers by carbonization at 800–2200 °C. Compared to carbon fibers from micron sized cellulose fibers, they found that graphitic structure was observed for CNFs at a relatively low temperature of 1500 °C, and that no obvious skin–core heterogeneity for fibers treated up to 2200 °C. The modulus of the CNF obtained at 2200 °C was ~100 GPa. Such mechanically strong CNFs are promising reinforcing fibers because of their large surface area contributing to the good interfacial adhesion and stress transfer in the composites [9]. Additionally, electrospun cellulose nanofiber based CNFs showed good electric conductivity, making them prospective electrochemical application materials [7, 10, 11]. Besides regenerated electrospun cellulose nanofibers were used as precursors, native cellulose nanofibers were good candidates too. For instance, amorphous CNFs of about 20 nm were fabricated by freeze-dried ball-milled cellulose nanofibers of ca. 50 nm at below 600 °C [12]. CNFs with diameter of 10–20 nm were synthesized using bacterial cellulose nanofibers as starting carbon sources, and were used as supportive scaffolds for biominerization [13].
Unlike the high and ordered graphitization of PAN-based carbon nanofibers [14], graphitic layers of cellulose-based carbon nanofibers had no preferred orientation [9]. These layers formed a network, and had no discontinuities at boundaries of the graphitic crystallites. This observation is different from the conventional graphitic layers presented in graphene materials [9, 15]. Thus, most authors thought they were glassy carbon. When cellulose is carbonized at a high temperature, the obtained solid carbon has a glass-like appearance and cannot be transformed into crystalline graphite even at 3000 °C and above, so the glassy carbon is a non-graphitizing carbon [16], with large amount of micropores and high content of amorphous carbon, which is quite different from the dense and highly crystallized CNFs using PAN and pitch as precursors. The glassy carbon displays high stability against heat, oxidizing gas, and acid. The oxidation rate of glassy carbon in oxygen, carbon dioxide, or water vapor is lower than that of any other carbons. With respect to the resistance to acid, it was demonstrated that normal graphite is reduced to powder by a mixture of concentrated sulfuric and nitric acids at room temperature, but glassy carbon is unaffected by such a treatment, even after several months [17]. The low reactivity, high hardness, impermeability, and biocompatibility of glassy carbon make it a good material for high temperature crucible, electrode [18, 19], and tissue engineering scaffold [20].
Different from the carbon nanofibers fabricated from cellulose nanofibers reported in the literatures [7], glassy carbon nanofibers (g-CNFs) from electrospun cellulose precursor nanofibers were prepared in this paper. The successful fabrication of g-CNFs is able to meet the resurging interest in the micro- and nano-devices of glassy carbon materials [21, 22]. The CelluNFs were stabilized at 400 °C and carbonized at 850 and 1400 °C into g-CNFs. The morphology and crystal structure of the precursor, stabilized and carbonized nanofibers were characterized by field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), selected-area electron diffraction (SAED), and Raman spectroscopy. The electrical conductivity of the g-CNFs was measured.
Experimental
Materials
Cellulose acetate (CA) (M n = 3.0 × 104 measured by GPC, 45.8 % acetyl content,) was purchased from Sigma-Aldrich, USA. Sodium hydroxide (NaOH), N,N-dimethylacetamide (DMAc), acetone, and absolute ethanol were bought from Sinopharm Chemical Reagent Co., Ltd. All chemicals were analytical grade and used without further purification.
Preparation of CelluNFs
CA powder was dissolved in a mixed solvent (acetone and DMAc, w/w = 2:1) to form a 15 wt% CA spinning solution. Then it was transferred into a 10-mL syringe with a blunt stainless needle. The CA nanofibers were fabricated on a lab setup electrospinning apparatus at 25 °C and 60 % relative humidity. A positively charged high-voltage power supply (DW-P303-IAC, Tianjin Dongwen High Voltage Plant, China) was connected to the needle tip with an inner diameter of 1 mm, while the ground electrode was connected to a plane collector covered with an aluminum foil. The voltage was 10 kV, and the distance between the needle tip and the collector was 20 cm. The feeding rate of the solution was set at 10 µL/min using a syringe pump (TS2-60, Longer Precision Pump Co. Ltd, Baoding, China). The as-spun CA nanofibers were dried in a vacuum oven at 40 °C for 8 h.
CA nanofibers were removed from aluminum foil and hydrolyzed in 0.1 mol/L NaOH/ethanol solution at 25 °C for 24 h, followed by washing with deionized water, until the water reaches neutral. The full hydrolysis of acetyl groups was confirmed by the absence of the band at 1752 cm−1 (–C = O) in its FTIR spectrum (not shown here). Afterward, the CelluNFs were air dried for 8 h, and then vacuum dried at 70 °C for 12 h.
Stabilization and carbonization of CelluNFs
The stabilization and carbonization of CelluNFs were performed by following the procedures described in the literatures [7, 9, 11, 23], with slight modification. In a typical procedure, the CelluNFs were treated in a GSL-1700X tube furnace (Hefei Kejing Materials Technology Co., LTD, China). A CelluNF mat was flatly placed in a crucible without tension, heated to 400 °C at a heating rate of 2 °C/min, then held for 1 h for stabilization. The stabilized nanofibers (SNFs) were subsequently heated at a rate of 2 °C/min to 850 °C, and held for 1 h. These processes were carried out under constant flow of high purity nitrogen. These carbonized nanofibers (C1NFs) were further carbonized in argon from 850 to 1400 °C at a heating rate of 5 °C/min, kept at 1400 °C for 1 h, then cooled down to room temperature. Such obtained CNFs were coded as C2NFs.
Characterization
The nanofiber morphology was observed on a field emission scanning electron microscope (FE-SEM, JSM-7500LV, JEOL) at 3 kV. Samples were platinum-coated before observation. The high-resolution TEM images and SAED patterns were obtained on Tecnai G2 F20 S-TWIN at an acceleration voltage of 200 kV. For TEM observation, the nanofibers were suspended in ethanol, sonicated for 20 min, and then a drop of suspension was placed on a copper grid. Raman spectrum was performed on an in via Microscopic Confocal Raman Spectrometer (Renishaw, England) with semiconductor laser light λ = 530 nm at 2 mW, and the integration time was 10 s.
The electrical conductivity (σ) of the g-CNF fibers was measured on their powder instead of a single g-CNF fiber or a CNF fibrous mat, because a CNF fiber is hard to be separated from the mat, and the mat is heterogeneous with large volume of pores. The CNF fibers were milled into powders in a mortar for 10 min. Using an apparatus for the preparation of KBr pellet for FTIR, 200 mg of CNF powders were pressed into circular pellet under 700 MPa for 2 min. In order to make an effective comparison, all samples were prepared in the same way. The σ was measured by CHI760c electrochemical workstation (Chenhua, Shanghai, China) at 30 °C. Five pellets were measured for each specimen. The σ was calculated by
where “R” is the electrical resistance in “ohms”, “A” is the area of the pellet in “cm2”, and “D” is the thickness of the pellet in “cm”.
Results and discussion
Morphology of nanofibers
In a preliminary study, we carbonized the CelluNFs at 850 °C directly, and found that the product was not in fiber morphology, instead it was in the form of particle. Thus, a sequential thermal treatment including stabilization and carbonization is necessary to retain the fiber morphology for the carbonized CelluNFs. The stabilization process involves decomposition and molecular rearrangement of the polymers [9], making them thermally more stable for the following carbonization process. It was reported that CelluNFs were effectively stabilized at 240 °C in air [9]. In our work, we stabilized the CelluNFs at 400 °C in nitrogen because relatively higher carbon yield and good carbon nanofiber morphology were obtained, as compared to CNFs made after stabilization at 200, 250, 300, and 350 °C. The topographies of CelluNF, SNF, C1NF, and C2NF are shown in Fig. 1. CelluNFs are continuous, relatively uniform in size, and randomly oriented into interwoven networks. After stabilization and carbonization (Fig. 1c–h), nanofibers are still continuous without noticeable fracture. The average diameters of CelluNF, SNF, C1NF, and C2NF are 230, 95, 82, and 45 nm, respectively, showing that the diameter of SNF (Fig. 1c, d) is substantially reduced to about 40 % of the size of precursor CelluNF. This is ascribed to the dehydration and thermal cleavage in cellulose molecular chains during the course of heating from room temperature to 400 °C. In this stage, large amount of gaseous products (H2O, CO or/and CO2) evolves from the pyrolysis of CelluNFs [6]. Compared to the straight morphology of CelluNF, SNF obviously becomes curled owning to shrinkage during stabilization. It was reported that major longitudinal shrinkage occurred in the stabilization stage, whereas that in the carbonization stage is slight [9]. The morphology of C1NFs carbonized at 850 °C (Fig. 1e, f) is very similar to that of SNFs. However, after carbonization at 1400 °C, nanofibers become even thinner and rougher, suggesting further carbonization and graphitization occurred (Fig. 1g, h).

SEM images of a CelluNFs, c SNFs, e C1NFs, and g C2NFs. b, d, f, h are the enlarged images of corresponding fibers
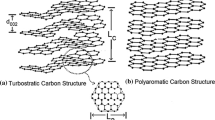
The C1NF and C2NF are further evaluated by TEM analysis. The sizes of C1NF and C2NF are ca. 71 and 50 nm, respectively. They display very different surface morphologies, i.e., the surface of C1NF is relatively smooth, while that of C2NF is very rough (Fig. 2). The HRTEM image of C1NF shows disordered and isotropic microstructure without obvious graphitic layers. It displays a granular texture, and tightly packing nanoparticles. The corresponding SAED patterns display two diffusion diffraction rings which are indexed to (110) and (101) lattice planes of rhombohedra amorphous carbon (JCPDS No. 26-1079). Obviously, C1NFs are amorphous carbon nanofibers. At a lower carbonization temperature of 600 °C, amorphous carbon nanofibers were also reported for the carbonization of the ball-milled cellulose nanofibers [12]. However, C2NFs, which was carbonized at 1400 °C, show ribbon-like curled single carbon layers throughout the sample. These layers are randomly stacked without any particular sequence, and are enclosed. Such a carbon layer is a fullerene-related structure, as suggested for the commercial glassy carbon SIGRADUR K&G [24]. Therefore, C2NFs are a typical glassy carbon with a turbostratic carbon structure. At a carbonization temperature higher than 2000 °C, it has been reported that curved graphitic walls typically containing entangled two to four layer planes are formed in the glassy carbon [9, 24]. Such entanglement is a resultant of crosslinking by sp3-bonded carbon atoms [25]. Due to the upper temperature limit of the furnace used in our work, we were unable to make glassy carbons at carbonization temperature over 1400 °C. But according to the transition trend shown in Fig. 2, it is highly possible to make a glassy carbon with more than two graphitic layers looping around each other without specific orientation [15].

TEM images of a, b C1NFs and c, d C2NFs. Insets are the corresponding SAED patterns
Raman spectroscopy
Figure 3 shows the Raman spectra of CelluNFs, SNFs, C1NFs, and C2NFs. For CelluNFs (Fig. 3a), the band at 1097 cm−1 is attributed to the vibration of C–O bond within the backbone of the cellulose chain. The 2895 cm−1 band is assigned to the –CH2– symmetric stretching, and the band at 1461 cm−1 is –CH2– bending mode which contains a very small proportion of C–OH bending. The 899 cm−1 is the methylene bending coordinates. 250–550 cm−1 are skeletal-bending modes involving the CCC, COC, OCC, and OCO internal coordinates. Small amount of methine bending (CCH and OCH) and skeletal stretching (C–O and C–C) also contributes in the region [26]. After stabilization, SNFs (Fig. 3b) show no cellulose bands but two faint bands at 1354 and 1572 cm−1, assigned to the respective D and G bands of carbon materials, which means that the structure of cellulose is changed with dehydration and thermal scission, resulting in increasing of carbon content in the course of stabilization. Subsequently, carbonization of the SNFs at 850 °C further alters their structure via thermal degradation and molecular rearrangement like aromatization, and the remaining product is mainly composed of carbon [6]. Thus, C1NFs show obvious D and G bands of carbon materials at 1295 and 1590 cm−1, respectively. The spectrum of C2NFs is similar to that of C1NFs, but showing sharper D and G bands centered at 1295 and 1596 cm−1, respectively. It is well known that D and G bands are both aroused from the vibration of sp2 bond; D band belongs to the A1g totally symmetric disorder mode occurring in sputtered or diamond-like carbons, while G band corresponds to the active E2g line of single-crystalline graphite [27]. It is worth noting that the D and G bands become sharper and narrower from SNFs to C1NFs and to C2NFs. These are attributable to the increasing aromatic structure coalesced by the sp2 carbons with increasing carbonization temperature [6, 9]. This is consistent with the TEM image showing more ribbon-like graphitic carbon layers in C2NF. Besides, the intensity of D band is related to the crystallite dimension, defect, and impurity of graphite, so the integrated intensity ratio of D to G bands (I D/I G) can be used to characterize the crystal structure and crystallite dimension [9, 28]. The value of I D/I G decreases with decreasing of defects or increasing of graphitization extent. The I D/I G values of C1NFs and C2NFs are 3.98 and 3.41, respectively. Such large I D/I G values indicate that C1NFs and C2NFs both have low graphite contents, and most of the carbon are amorphous even though C2NFs exhibit a little higher graphite content than C1NFs. Interestingly, with increasing carbonization temperature the D band is 4 cm−1 blue-shifted while the G band is 4 cm−1 red-shifted. These are ascribed to the dehydrogenation-induced Fermi level shift during carbonization [29].

Raman spectra of a CelluNFs, b SNFs, c C1NFs, and d C2NFs
Electrical conductivity (σ) of g-CNF
The σ of cellulose is 10−8 S/cm. Thermal treatment may greatly increase it to 10−4 S/cm when treated at 600 °C [6], and to 3.8 S/cm for cellulose-based carbon nanofibers treated at 800 °C [7]. The g-CNFs derived from cellulose nanofibers are fragile. It is hard to measure the electrical conductivity of a single g-CNF because of difficult separation it from the mat. Meanwhile, the reproducibility or reliability of the σ of the g-CNF mat is poor because of the heterogeneous structure of the g-CNF mat with high pore volume (>90 %). Therefore, the σ of g-CNF powder was determined in this work. The σ is 8.7 ± 0.3 and 93.5 ± 1.4 S/cm for g-C1NF and g-C2NF, respectively. Obviously, the conductivity is closely related to the level of disorder in g-CNF. TEM image demonstrates that highly disordered amorphous carbon are present in the g-C1NF carbonized at 800 °C, whereas much of the disorder is removed and entangled ribbon-like graphitic carbon layer networks are formed in the g-C2NF carbonized at 1400 °C. Raman spectra also quantitatively suggest that more graphitic crystallites are produced in the g-C2NF than that in the g-C1NF. The more ordered microstructure in g-C2NF leads to much improved conductivity. The σ of g-C1NF is in the range of amorphous carbon, whereas the σ of g-C2NF is almost in the same order of other polymer-based glassy carbons. For example, furfuryl alcohol based glassy carbon nanowire carbonized at 1500 °C had σ of 45 S/cm [22], and phenol formaldehyde based glassy carbon carbonized at 1400 °C showed σ of 179.3 S/cm [30]. The g-C2NF with good electrical conductivity may find potential application in the carbon based advanced electrochemical materials.
Conclusion
Glassy carbon nanofibers were fabricated via stabilization and the following carbonization of electrospun cellulose nanofibers. Stabilization and carbonization did not change the integrity of nanofiber morphology, but the average diameter of nanofibers greatly reduced from 230 to 45 nm. The reduction of nanofiber diameter was caused by the pyrolysis, depolymerization, aromatization, carbonization, and graphitization processes. C1NFs and C2NFs both showed strong D and G bands of carbon, and the value of I D/I G was 3.98 and 3.41, respectively. The large I D/I G value confirmed that C1NFs and C2NFs were almost amorphous carbons, which was consistent with the glassy carbon structure demonstrated by the TEM images. The glassy carbon nanofibers showed random network of ribbons composed of stacked carbon layers. The electrical conductivity of g-CNF was related to its microstructure, which greatly depended on the carbonization temperature. The electrical conductivities of g-C1NF and g-C2NF were 8.7 and 93.5 S/cm, respectively. Because of the disordered graphitic structure in the g-C2NF, its electrical conductivity was much higher than that of g-C1NF, but much lesser than that of graphene (~105 S/cm, parallel to the basal plane).
References
Maiyalagan T, Scott K (2010) Performance of carbon nanofiber supported Pd–Ni catalysts for electro-oxidation of ethanol in alkaline medium. J Power Sources 195:5246–5251
Miao J, Miyauchi M, Simmons TJ, Dordick JS, Linhardt RJ (2010) Electrospinning of nanomaterials and applications in electronic components and devices. J Nanosci Nanotechnol 10:5507–5519
Kim BJ, Lee YS, Park SJ (2008) A study on the hydrogen storage capacity of Ni-plated porous carbon nanofibers. Int J Hydrogen Energy 33:4112–4115
Wu MY, Wang QY, Liu XQ, Liu HQ (2012) Biomimetic synthesis and characterization of carbon nanofiber/hydroxyapatite composite scaffolds. Carbon 51:335–345
Marchessault RH (2011) All things cellulose: a personal account of some historic events. Cellulose 18:1377–1379
Dumanlı AG, Windle AH (2012) Carbon fibres from cellulosic precursors: a review. J Mater Sci 47:4236–4250. doi:10.1007/s10853-011-6081-8
Kuzmenko V, Naboka O, Gatenholm P, Enoksson P (2014) Ammonium chloride promoted synthesis of carbon nanofibers from electrospun cellulose acetate. Carbon 67:694–703
Kong K, Deng L, Kinloch IA, Young RJ, Eichhorn SJ (2012) Production of carbon fibres from a pyrolysed and graphitised liquid crystalline cellulose fibre precursor. J Mater Sci 47:5402–5410. doi:10.1007/s10853-012-6426-y
Deng L, Young RJ, Kinloch IA, Zhu Y, Eichhorn SJ (2013) Carbon nanofibres produced from electrospun cellulose nanofibres. Carbon 58:66–75
Naboka O, Rodriguez K, Toomadj AF et al (2012) Carbon nanofibers synthesized from electrospun cellulose for advanced material applications. Mater Sci Forum 730–732:903–908
Deng L, Young RJ, Kinloch IA et al (2013) Supercapacitance from cellulose and carbon nanotube nanocomposite fibers. ACS Appl Mater Interface 5:9983–9990
Jazaeri E, Zhang L, Wang X, Tsuzuki T (2011) Fabrication of carbon nanofiber by pyrolysis of freeze-dried cellulose nanofiber. Cellulose 18:1481–1485
Wan Y, Zuo G, Yu F, Huang Y, Ren K, Luo H (2011) Preparation and mineralization of three-dimensional carbon nanofibers from bacterial cellulose as potential scaffolds for bone tissue engineering. Surf Coat Technol 205:2938–2946
Zhou Z, Lai C, Zhang L et al (2009) Development of carbon nanofibers from aligned electrospun polyacrylonitrile nanofiber bundles and characterization of their microstructural, electrical, and mechanical properties. Polymer 50:2999–3006
Jenkins GM, Kawamura K (1971) Structure of glassy carbon. Nature 231:175–176
Kaburagi Y, Hosoya K, Yoshida A, Hishiyama Y (2005) Thin graphite skin on glass-like carbon fiber prepared at high temperature from cellulose fiber. Carbon 43:2817–2819
Cowlard FC, Lewis JC (1967) Vitreous carbon—A new form of carbon. J Mater Sci 2:507–512. doi:10.1007/BF00752216
Rajith L, Jissy A, Kumar KG, Datta A (2011) Mechanistic study for the facile oxidation of trimethoprim on a manganese porphyrin incorporated glassy carbon electrode. J Phys Chem C 115:21858–21864
Park S, Park DW, Yang CS et al (2011) Vertically aligned carbon nanotube electrodes directly grown on a glassy carbon electrode. ACS Nano 5:7061–7068
Lehr J, Williamson BE, Downard AJ (2011) Spontaneous grafting of nitrophenyl groups to planar glassy carbon substrates: evidence for two mechanisms. J Phys Chem C 115:6629–6634
Du R, Ssenyange S, Aktary M, McDermott MT (2009) Fabrication and characterization of graphitic carbon nanostructures with controllable size, shape, and position. Small 5:1162–1168
Lentz C, Samuel B, Foley H, Haque M (2011) Synthesis and characterization of glassy carbon nanowires. J Nanomater 2011:9
Ishida O, Kim DY, Kuga S, Nishiyama Y, Brown JRM (2004) Microfibrillar carbon from native cellulose. Cellulose 11:475–480
Harris PFJ (2004) Fullerene-related structure of commercial glassy carbons. Philos Mag 84:3159–3167
Saxena RR, Bragg RH (1978) Electrical conduction in glassy carbon. J Non-Crystalline Solids 28:45–60
Wiley JH, Atalla RH (1987) Band assignments in the Raman spectra of celluloses. Carbohydr Res 160:113–129
Polarz S, Smarsly B, Schattka JH (2002) Hierachical porous carbon structures from cellulose acetate fibers. Chem Mater 14:2940–2945
Jaiswal M, Yi Xuan Lim CH, Bao Q, Toh CT, Loh KP, Özyilmaz B (2011) Controlled hydrogenation of graphene sheets and nanoribbons. ACS Nano 5:888–896
Ryu S, Han MY, Maultzsch J et al (2008) Reversible basal plane hydrogenation of graphene. Nano Lett 8:4597–4602
Soukup L, Gregora I, Jastrabik L, Koňáková A (1992) Raman spectra and electrical conductivity of glassy carbon. Mater Sci Eng, B 11:355–357
Acknowledgements
This work is supported by the Research Fund for the Doctoral Program of Higher Education of China (No. 20123503110003), the National Natural Science Foundation of China (No. 50973019), the National Basic Research Program of China (2010CB732203), and the Education Department of Fujian Province (JA13079).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, Y., Qin, W., Wang, Q. et al. Glassy carbon nanofibers from electrospun cellulose nanofiber. J Mater Sci 50, 563–569 (2015). https://doi.org/10.1007/s10853-014-8612-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8612-6