
Abstract
In this study, we attempt to ascertain the accuracy of the structures determined using our previously developed method and hence the accuracy of our method. In the present report, we have taken the guest molecule cetirizine (CTZ) and the host molecules are α-cyclodextrin (α-CD) and β-cyclodextrin (β-CD). Structures with good accuracy were elucidated using a productive fusion of experimental and computational methods. We performed molecular mechanics studies (MM) in light of experimental ROESY studies, followed by molecular dynamics studies (MD). The results from these studies were analyzed using quantitative ROESY analysis to determine the final accurate structures of the complexes. The accuracy of these structures was tested using density functional theory (DFT) that is an accurate method for structure determination. DFT studies were carried out using the functionals B3LYP and M06L with def-TZVP basis set and similarly quantitative ROESY analysis was performed for the obtained structures. The ROESY intensities of the structures obtained from MM and MD studies, were compared with ROESY intensities of the structures obtained from DFT studies. Calculated ROESY intensities of the structures obtained from B3LYP functional are comparable, with intensities of structures obtained from MM and MD studies, but M06L functional showed poor results. In addition to the accuracy of MM and MD studies, low computational cost and less time input make it good method for structural studies for CD inclusion complexes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biological systems such as proteins, lipids, nucleic acids etc., are fundamental for the organization of a cell. Their interaction with each other and extracellular particles controls various aspects of life. These biomolecules enable systematic functioning of various biological processes inside the cell, in a well-defined manner, to maintain the life of an organism. The basic functioning of these systems can be understood by supramolecular chemistry [1]. It is the study of multimolecular systems formed as a result of non-covalent interactions. It exhibits several important concepts like molecular recognition, chiral recognition, molecular self-assembly, etc. Molecular and chiral recognition are of great importance, as they form the basis of supramolecular chemistry. Molecular recognition is the specific interaction between two or more molecules that exhibit complementarity in size, geometries, and functional groups. Chiral recognition is the chemical interaction by which a receptor molecule recognizes a particular enantiomer of the substrate. The detailed study of weak non-covalent interactions responsible for molecular and chiral recognition is crucial to understand various biological processes. Since biological systems are large in size for practical study so to understand these systems suitable models of intermediate sizes mimicking the biomolecules are required. Cyclodextrin (CD) inclusion complexes are considered as good models by chemists to explore these non-covalent interactions [2]. CDs have found their use as one of the best hosts because of their ability to form a series of water-soluble host molecules. Therefore, to understand the molecular and chiral recognition phenomena, structures of the CD inclusion complexes need to be explored [3].
The structural studies of CD inclusion complexes are under investigation worldwide. The highfield shift in the 1H-NMR signals of CD cavity protons was the first direct evidence for the inclusion of guest molecule into CD cavity [4]. 1H-NMR titration data have been used to obtain stoichiometry and association constant of CD inclusion complexes [5]. Advancements in 1H-NMR spectroscopy i.e. 2D-ROESY helped in identifying the part of the guest encapsulated, guest orientation, mode of entry and depth of inclusion into the CD cavity [6]. In light of ROESY contacts, computational studies like MM, MD, etc. were performed and the minimum energy structure was usually considered as the final structure. But the atom accuracy of these structures was probably never tested. We also have developed a method of quantitative ROESY, where the calculated ROESY intensities are compared with experimental ROESY intensities, for obtained computational models of CD inclusion complexes [2]. Using this method structures of CD inclusion complexes has been achieved with good accuracy [7].
Butts and coworkers used experimental NOE intensities to determine interproton distances, in a rigid organic molecule strychnine [8, 9]. These interproton distances were found to fit extremely well with structures, determined by crystallographic and DFT calculations. This method was also successfully applied for the structural determination of small flexible molecules like 4-propylaniline with high accuracy [10]. Later on, they developed method for structure determination of conicasterol F, where interproton distances were obtained from quantitative analysis of ROEs and compared with structures obtained from DFT calculations [11]. This method was found to provide highly atom-accurate structures.
We have used the strategy of Butts and coworkers in CD inclusion complexes for the first time, and highly atom accurate structures have been determined by this method. We perform MM studies, where the guest molecule is placed manually inside the CD cavity at different depths, followed by energy minimization. Based on MM, the MD studies are carried out to simulate the inclusion process. The interproton distances for minimum energy conformations from MM and MD frames are used to calculate ROESY intensities. Then these calculated ROESY intensities are compared with experimental ROESY intensities. The structure for which calculated ROESY intensities better match experimental ROESY intensities, is proposed as the final structure [2]. Since, final structure of MD simulation depends on the initial frame, so several MDs need to be performed, until calculated ROESY intensities for minimum energy frame match experimental ROESY intensities, to obtain atom accurate structures. The atom accuracy of the structures proposed by our method depends on the quality of ROESY spectrum, which in turn depends on mixing time. A good quality ROESY spectrum is recorded at shorter mixing time, and using this highly atom accurate structures have been proposed by our method. But the problem is it requires large experimental time and is costly. Therefore, ROESY spectrum is recorded at long mixing time, to decrease cost and experimental time. The ROESY spectrum recorded at long mixing time shows the overlapping of peaks, resulting into the poor quality of ROESY [5]. We are interested in finding the atom accuracy of proposed structures, using poor quality ROESY. For this purpose, we need to compare results obtained by our method with the results obtained from a well-established and standard method DFT. B3LYP functional is hybrid functional and is considered as standard level of DFT theory, because it provides reliable results [12].
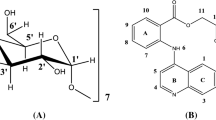
In this article we have studied the complexation of cetirizine (CTZ) with α-CD and β-CD (Fig. 1). The initial characterization and inclusion process of CTZ with CDs have been already studied by us as well as by others [6, 7, 13, 14]. Initially the structures of inclusion complexes were obtained using ROESY and MD by quantitative analysis. Then the structures were obtained by a well-established method DFT using B3LYP and M06L level of the theory and def-TVZP as the basis set. The structures of inclusion complexes obtained by our method were compared with the structure obtained by DFT.

Chemical structure of cetirizine, α-CD and β-CD

2D structure showing hollow cavity of CDs
Experimental
2D-ROESY (Figs. 3 and 4) and COSY spectra for CTZ/α-CD and CTZ/β-CD mixture (1:1 ratio) were recorded in D2O on a JEOL 500 MHz instrument at room temperature. COSY spectrum was recorded utilizing 1:1 pulsed field gradient with a relaxation delay and repetition time of 1.5 s and 1.609 s respectively. 2D ROESY spectra were recorded under spin lock conditions with mixing time of 0.25 s, relaxation time of 1.5 s and repetition time of 1.609 s. Molecular mechanics (MM) and molecular dynamics (MD) studies were executed using Allinger`s force field at room temperature in CS Chem3D Pro (Cambridge Soft Corp.) in the vapor phase [15]. CTZ was drawn in CS ChemDraw Pro 2D and converted in C3D format by CS Chem3D Pro. This was further minimized using Aligners force field (MM2) and was used for MM and MD studies. Published neutron diffraction coordinates of hydrated α- and β-CD were used, after removal of water coordinates [16, 17]. DFT studies were performed in Orca 4.1.1 version [18]. AvogadroOrca4.1 was used to generate input to orca and as visualization tool to optimized structures. Notepad+ + was used to read input to Orca and output from Orca. The minimum energy frames obtained from MM studies were converted into pdb format, whose coordinates were taken as input to DFT studies. DFT studies were performed at B3LYP and M06L level of theory utilizing def-TZVP as basis set.

ROESY spectrum of CTZ/α-CD mixture (1:1 ratio) with expansion of the region showing interaction between cavity protons and aromatic protons of the guest

ROESY spectrum of CTZ/β-CD mixture (1:1 ratio) with expansion of the region showing interaction between cavity protons and aromatic protons of the guest on both sides of diagonal
Results and discussion
The 1H–1H ROESY spectra
The CDs are composed of d-glucopyranose units joined by α-1, 4-linkage with each other in cyclic manner. The number of glucopyranose units in α-, β-, and γ-CDs are six, seven and eight respectively. Each glucopyranose unit consists of six different types of hydrogens directly attached to its carbons. The inner cavities of CDs consist of a band of H-3′ protons situated near the wider end, and a band of H-5′ protons situated towards the narrow end, whose numbers are equivalent to number of D-glucopyranose units present in CD. The H-1′, H-2′ and H-4′ lie on exterior of CD cavity, where H-2′ is closer to the wider end, below which are situated H-1′ and H-4′. The H-6′ protons are present on the narrow rim of the cavity. There are two secondary –OH groups at wider end and one primary –OH group at narrower end of the cavity, making its exterior hydrophilic. However, the interior of the cavity is less hydrophilic than exterior surface, hence making it suitable to accommodate hydrophobic guests into it. Figure 2 shows the position of α- and β-CD protons in relation with the wide and narrow ends of the cavity. The cavities of α-, β- and γ-CD preferably bind with aromatic guests as they have diameters large enough to accommodate aromatic rings. α-CD can also form complexes very well with long-chain fatty acids, particularly the polyunsaturated ones, depending on the geometry of the fatty acids. Experimentally, it can be shown by 2D-ROESY that whether guest is either included or not into CD cavity.
ROESY is a type of 2D spectroscopy where cross peaks arise due to space interaction between neighboring nuclei, close to each other. Analysis of these cross peaks between the guest and CD cavity protons in the ROESY spectrum provides information on the depth of inclusion, mode of entry and part of the guest encapsulated into the cavity. The α-CD consist of six glucose units and hence six protons of H-3′ and H-5′ in each ring. Similarly, β-CD consist of seven glucose units and hence seven protons of H-3′ and H-5′ in each ring. A qualitative analysis of a ROESY contact observed between a guest proton and any one of the cavity protons, does not provide any information about their closeness i.e. the distance between that particular guest proton and anyone of those cavity protons. A minor change in the position of guest leads to change in all interproton distance between guest and CD cavity. Therefore, analysis of these interactions quantitatively i.e. by relating interproton distances with peak intensity is necessary to obtain highly atom accurate structures.
ROESY spectrum of CTZ/α-CD mixture (Fig. 3) shows a single large peak of interaction between aromatic protons of guest and CD cavity protons on one side of diagonal, and two peaks on other side of diagonal. The cross peak on left side of diagonal shows that only chlorophenyl ring of CTZ forms complex with α-CD. The cross peaks on the right side of the diagonal shows again the interaction of aromatic protons with cavity protons confirming inclusion complex formation. Moreover, relative peak analysis of aromatic protons with CD cavity protons shows that the guest has entered into cavity from the wider end. The cavity size of α-CD is large enough to accommodate one aromatic ring inside the cavity. Therefore, it was considered from our previous studies that host is to guest ratio is 1:1. The analysis of NMR signals was done using COSY spectrum of CTZ/ α-CD mixture (1:1 ratio).
The analysis of the ROESY spectrum (figure) of CTZ/β-CD mixture (1:1) shows cross correlation peaks between the guest and β-CD cavity protons (H-3′, H-5′). The ROESY spectrum of CTZ/β-CD mixture (Fig. 4) shows two peaks of interaction, on each side of diagonal, between CD cavity protons and aromatic protons of the guest. The two peaks on the left side of diagonal show entry of both phenyl and chlorophenyl rings into CD cavity, showing formation of two inclusion complexes between CTZ and β-CD. It is clear from relative peak intensity analysis that, ROE of phenyl ring protons is large as compared to chlorophenyl ring protons. Similarly, the cross peaks on the other side of the diagonal also confirms the complexation of both aromatic rings of CTZ with β-CD cavity. The ROESY spectrum analysis also shows that guest has entered into β-CD cavity from wider end. The NMR peak analysis and ratio of guest is to host was taken as 1:1 according to previous literature.
Computational studies
For the complexation of CTZ with both α-CD and β-CD, molecular mechanics (MM) and molecular dynamics (MD) simulations were carried out in CS Chem3D Pro (Cambridge soft corp.), using Allinger’s force field in the vapor phase [15] at room temperature. The structures of all inclusion complexes i.e. CTZ/α-CD and CTZ/ β-CD were drawn by minimizing their geometries to root mean square gradient of 0.1 kcal/mol. During the simulations performed for the inclusion complexes under study, host molecules (α-CD and β-CD) were kept static and guest molecule (CTZ) was allowed to move because CDs adopt more symmetrical geometry in complexes of solid as well as solution state. DFT calculations were carried in ORCA4.1.1 [18] at B3LYP and M06L levels of the theory and basis set def-TZVP. These studies were performed in vaccum as there are no water molecules present in CD cavity in complexed state. Notepad++ was used to read input to orca and output from orca. AvogadroOrca4.1 was used to visualize the output structures of inclusion complexes formed from orca.
Molecular mechanics (MM)
MM studies provide local minima and have been used to establish the minimum energy structures of the inclusion complexes formed. In the MM studies, the guest molecule was placed manually inside the host cavity followed by energy minimization. Using CS Chem 3D Pro, MM2 energy minimizations were performed for all the inclusion complexes of CTZ/α-CD and CTZ/β-CD. We have used different depths (Fig. 5) and different modes (Fig. 6) for placing the aromatic rings perpendicular to the diameter of the cavity for both α-CD as well as β-CD. All the above energy minimization processes were done at three different depths (Fig. 5) from both the narrow side (NS) and wide side (WS). The three different depths are named as Narrow Surface (N1), Narrow center (N2) and Narrow Bottom (N3) on narrow side. Similarly, on the wide side these are named as Wide Surface (W1), Wide center (W2) and Wide Bottom (W3). The three different modes used are shown in Fig. 6, where one mode shows entry of para carbon of aromatic ring into cavity and other two modes show entry of aromatic ring by side of bonds alternate to para carbon.

Different depths at which molecular mechanics studies were done

Different modes of study used in molecular mechanics
The binding energy (B.E.) for the inclusion complex formed was calculated using formula
Higher the negative value of the binding energy more stable is the complex and proves the spontaneity of inclusion process [19].
In case of α-CD, examining the ROESY spectra revealed that only the chlorophenyl ring of CTZ forms an inclusion complex and hence MM2 studies were performed only for the chlorophenyl ring of CTZ. Energy minimizations were performed from both narrow (N1, N2, N3) and wide side (W1, W2, W3) of the CD cavity as well as in different modes. The minimum energy structure hence obtained was from mode 1 with the W2 inclusion depth. The chlorophenyl ring was inserted from the wider end and placed at the center of the cavity with the entry of the chlorine atom perpendicular to the Z-axis of the cavity. The chlorophenyl ring tilted slightly after MM calculations with an energy of 80.611 kcal/mol (B.E. = − 27.076 kcal/mol). The top and side view for this minimum energy conformation is shown in Fig. 7. The binding energy values for different possible modes are shown in Table 1.

Lowest energy structure of CTZ/α-CD from MM studies
In the CTZ/ β-CD complex, the energy minimization was performed for both chlorophenyl as well as phenyl rings, using different modes and different depths since, ROESY spectrum confirmed complex formation by both chlorophenyl and phenyl rings. Further, it was investigated whether the rings of CTZ form a single complex with simultaneous inclusion of both the rings or two separate complexes of the rings with β-CD were formed. We manually placed both rings into the cavity at the same time and performed MM study. Energy minimization resulted in complete removal of both the rings from the cavity, which shows that the phenyl and chlorophenyl rings form two separate complexes with β-CD rather than a single complex with both the rings included together. Therefore, MM studies were performed for both the aromatic rings of CTZ with CD individually.
The minimum energy conformation of chlorophenyl ring with β-CD was found in depth i.e. W3 and mode 2 with steric energy 89.481 kcal/mol (B.E. = − 31.197 kcal/mol). Similarly, minimum energy conformation of phenyl ring with β-CD was found at bottom of cavity by placing phenyl ring of CTZ in mode 1 (W3) having steric energy 86.880 kcal/mol (B.E. = − 33.798 kcal/mol), where ring gave a little tilted conformation after energy minimization. The top and side views for both the minimum energy complexes are shown in Fig. 8. The energy values for different possible modes are shown in Table 2. The negative value of the binding energy shows that the complexation process is spontaneous.

Lowest energy structure of CTZ/β-CD complexes from MM studies A) Phenyl/CTZ complex B) Chlorophenyl/CTZ complex
Molecular dynamics (MD)
The MD simulation study initially used to simulate small molecules, has now advanced as an important tool, and is used in routine studies of larger and complex molecules. The inclusion process of CD inclusion complexes is studied on the large scale, to provide atomic level representation of guests inside the CD cavity. To further confirm the results obtained from the MM studies and to obtain more accurate representation of the structure of the complexes formed, MD simulations were carried out in light of ROESY results. From ROESY spectra it was found that in case of CTZ/α-CD complex only the chlorophenyl ring of CTZ forms complex α-CD cavity whereas, both phenyl and chlorophenyl rings of CTZ form complex with β-CD. MD simulations were performed for both the rings of CTZ with β-CD and only for the chlorophenyl ring with α-CD. The MD simulation study was set to terminate after 10,000 equilibrium steps resulting into trajectory of 1000 frames, with a step interval of 1.0 fs and frame interval of 10 fs for each run. The geometries of these structures were minimized to root mean square gradient of 0.1 kcalmol/Å. During the simulation process the guest molecule (CTZ) was allowed to move freely whereas the host molecules (α- and β-CD) were kept static to preserve symmetry of the cavity. The MD simulation studies were carried in Chem3D Pro using Alligner’s force field [15] in vapor phase [16]. Parameters for explicit solvation modeling of CD complexes have not been parameterized fully, and therefore, modeling of CD complexes is mostly performed in vacuum. Therefore, MD simulations were performed in vacuum. Several MD simulation studies were done for each inclusion complexation, of which the MD run whose minimum energy conformer better matched the ROESY peak intensities was considered as final structure.
In CTZ/α-CD complex, the MD studies were performed from wide side of the cavity as confirmed from MM studies. Further, MD studies were also performed from the narrow side of the cavity, the energy-time plot of which shows that wide side entry was more favorable on the basis of steric energy. In MD studies of CTZ/α-CD from wide side the chlorophenyl ring was placed on the surface of α-CD cavity, perpendicular to diameter of cavity, in such a way that chlorine of CTZ was in level of H3′ cavity protons. As the command for MD studies was given, the molecule showed movement towards the bottom of cavity till 1600 fs. The chlorophenyl ring stayed at the same position up to 2000 fs showing vibration however, during this time the other part i.e. phenyl and aliphatic of CTZ showed a large movement. After 2000 fs the chlorophenyl ring moved back to center of the cavity and stayed there till 3140 fs. The ring then showed surface to bottom motion with minimum energy frame at 5790 fs. After 6750 fs, the ring gets tilted showing the entry through the side of the bond. During whole run aliphatic part of CTZ have also shown a large movement. The energy time plot, minimum energy conformation (frame 579), and few frames of MD simulation are shown in Figs. 9, 10, and S5. respectively.

Energy-time plot of MD run for complexation between α-CD and Chlorophenyl ring of CTZ

Side and top view of least energy frame of MD simulation between Chlorophenyl ring and α-CD
In case of CTZ/β-CD complexation process, several MD simulations were performed with different possibilities of guest orientation into the host cavity. In order to confirm whether the phenyl and chlorophenyl rings form a single or two different inclusion complexes, MD simulations were performed keeping the CTZ molecule such that both rings were included in the cavity together. During the run, the rings moved out of the cavity each time when MD was initiated with inclusion of both the rings. This pointed to the formation of two different inclusion complexes by the aromatic rings of CTZ. Next MD simulations were performed for the phenyl and chlorophenyl rings separately from the wide as well as narrow side of the cavity. On the basis of energy, wide side entry was found to be more favorable than narrow side entry in both the complexes formed by the aromatic rings of CTZ. The MD simulations for which the minimum energy structure matches best with ROESY interactions have been reported. MD simulations were performed for both the aromatic rings from wider side of the cavity, placing the rings at the surface of the cavity in their respective complexes.
In phenyl/β-CD complexation the phenyl ring was placed perpendicular on to the surface of CD cavity as shown in frame 1 (Fig. S6) and then MD studies were initiated. In the initial frame para-hydrogen of phenyl ring was kept at depth of H3′ cavity protons and ortho hydrogens on the surface of cavity. During simulation the molecule showed the movement towards the bottom of the cavity till 1320 fs and returned back to the surface of the cavity after 2070 fs showing minimum energy frame after 2617 fs. After 2070 fs the phenyl ring with remaining part of CTZ outside cavity showed movement in all possible orientation. Of all the frames, the minimum energy frame (frame 261) with steric energy 86.004 kcal/mol (B.E. = − 34.673 kcal/mol) was considered as final structure (Fig. 11, 12). The energy-time plot and few snapshots for this MD simulation are shown in Fig. 13, and S6 respectively.

Energy-time plot of MD run for complexation between β-CD and phenyl ring of CTZ

Side and top view of least energy frame of MD simulation between phenyl ring and β-CD
In the chlorophenyl/β-CD simulation studies chlorophenyl ring was placed near the cavity such that chlorine was present at the center of cavity and meta-hydrogens on the surface of the cavity (Fig. S7). The chlorophenyl ring was placed perpendicular to the diameter of cavity. As the MD simulation studies were commenced Chlorophenyl ring of CTZ molecule showed the movement towards the bottom of the cavity till 880 fs and started rotation at same position up to the 1480 fs. The ring then returned back to the surface of the cavity after 2200 fs where it got slightly tilted. The chlorophenyl ring then again showed movement towards the bottom of cavity where chlorine atom was seen out of narrow end at 4510 fs. After this, the chlorophenyl ring showed movement between center and bottom of CD cavity, showing minimum energy frame after 4580 fs (frame 458). During whole simulation process aliphatic part of CTZ as well as phenyl ring of CTZ also showed a large movement. The minimum energy frame (frame 458) with steric energy 85.926 kcal/mol (B.E. = − 34.563 kcal/mol) was considered as final structure (Fig. 14) of this complex formed. The energy-time plot for this MD simulation is shown in Fig. 13 with few MD snapshots in Fig. S7.

Energy-time plot of MD run for complexation between β-CD and chlorophenyl ring of CTZ
DFT studies
DFT has found various important applications in a large number of multi electron systems [20]. Geometry optimization of the CD inclusion complexes formed by CTZ with α-CD and β-CD was performed. In case of complex between CTZ and α-CD, the geometry optimization was done only for chlorophenyl ring, however for CTZ complexation with β-CD, DFT based geometry optimization was done for both chlorophenyl as well as phenyl ring. DFT is an expensive and highly CPU demanding method [21]. Therefore, DFT based geometry optimization was not possible from both the narrow as well as wider cavity end of the CD cavity. So, to make the calculations easy, geometry optimization was performed only from the wider end as was obtained from ROESY, MM and MD results. DFT studies were performed using the B3LYP and M06L functionals and def-TZVP was set as the basis set for both the functionals. M06L, a GCA functional was used because it is less expensive and less time taking method as compared to a hybrid functional B3LYP. B3LYP is a hybrid functional and is also considered as a standard functional, used widely for geometry optimization [20]. We have already got highly atom accurate results in CTZ/γ-CD complexation using this functional. Here, we have studied the complexation of CTZ with α- and β-CD. The coordinates of the minimum energy structures from MM were used as input to DFT studies. The Geometry optimization was performed in ORCA 4.1.1 [18]. The notepad+ + was used to read input to ORCA and output from the ORCA. The input to ORCA was generated using AvogadroORCA4.1.
In case of CTZ and α-CD complexation the geometry optimization was completed in 136 cycles using B3LYP and 190 Cycles with M06L functional. The formation of intermolecular Hydrogen bonds (Table 3) was found within standard range of bond angles and bond lengths. Usually it is seen that intermolecular H-bonds having smaller bond lengths are strong and these stabilize the molecules by keeping host–guest intact. The donor–acceptor distance in strong H-bonds is less than 2.55A0 for O–H–O systems [22]. The formation of these intermolecular H-bonds is considered as one of the force responsible for spontaneity in formation of inclusion complexes [7]. In CTZ/α-CD, four H-bonds were seen for the structure (Fig. 15) optimized by B3LYP functional and three H-bonds in structure optimized by M06L functional. The binding energy values for geometry optimized structures of Chlorophenyl/α-CD complex using B3LYP functional (− 30.168 kcal/mol) and a Minnesota functional M06L (B.E. = − 28.661 kcal mol) showed that the process of complexation is spontaneous.

Side and top view of least energy frame of MD simulation between chlorophenyl ring and β-CD
To show the H-bonds, the numbering in CTZ/ α-CD inclusion complex was done separately for α-CD and CTZ. α-CD was numbered using superscript (ʹ) and CTZ was numbered without superscript. In α-CD hydrogens were numbered using natural numbers (1, 2, 3) and oxygens were numbered as lowercase alphabets (a, b, c) with superscript (ʹ). In case of CTZ hydrogens were numbered using natural numbers (1, 2, 3) and oxygens were numbered as lowercase alphabets (a, b, c) without Superscript. The numbering in α-CD was difficult because it is large in size however, in case of CTZ was easy since it is relatively smaller. To simplify numbering in α-CD, numbering was done for only one of the six glucopyranose units since, all the six units are identical and were named as ring-A, ring-B and so on to ring-F. The numbering for both CTZ and α-CD is shown in Fig. 1.
In CTZ and β-CD complexation, geometry optimization using B3LYP level of the theory was completed after 225 optimization cycles in case of phenyl ring and 233 optimization cycles in case of chlorophenyl ring. Both the complexes of CTZ with β-CD have shown the formation of intermolecular H-bonds (Table 4). In Chlorophenyl ring/β-CD inclusion complex, four intermolecular H-bonds were formed and in phenyl ring/β-CD four intermolecular H-bonds were formed. The table shows the data of bond lengths, bond angles and bond strength of the intermolecular H-bonds formed in CTZ/β-CD complexes whose structure (Fig. 16) was optimized at B3LYP level of the theory. The binding energy values for chlorophenyl and phenyl/β-CD complexes whose structures were optimized using B3LYP functional, was found to be − 29.011 kcal/mol and − 32.660 kcal/mol respectively. These negative values of binding energies show that complexation process was spontaneous for both the complexes formed between CTZ and β-CD. To show the H-bonds in CTZ/β-CD complexes numbering was done in the same order as shown in CTZ/α-CD complex except β-CD contains seven glucopyranose units which were numbered from Ring A to Ring G. The numbering for CTZ and β-CD is shown in Fig. 1. It is usually accepted that smaller hydrogen bonds are stronger and as the bond length increases the bond strength decreases. In stronger H-bonds the donor–acceptor distance is up to 2.4–2.5 A0 for O–H–O systems and 2.5–2.6 A0 N–H–O systems [22]. Further H-bonds having bond lengths greater than 2.7 A0 are and below 3.8 A0 are considered to be of intermediate strength. The H-bond having bond lengths above 3.8 A0 are considered to be weak bonds [7].

Side and top view of the Chlorophenyl/α-CD inclusion complex structure optimized by DFT, a Using B3LYP, b Using M06L
The geometry optimization of CTZ/β-CD using M06L level of the theory was completed in 190 cycles for Chlorophenyl ring/β-CD and 324 cycles for phenyl ring. Both the complexes have shown the formation of intermolecular H-bonds, which are responsible for holding host and guest molecules together. Geometry optimized structures (Fig. 17) using M06L level of the theory showed that, three intermolecular H-bonds were formed in Chlorophenyl ring/β-CD complex and six intermolecular H-bonds were formed in phenyl/β-CD complex (Table 5). The negative values of binding energies for chlorophenyl/β-CD complex (B.E. = −28.661 kcal/mol) and phenyl/β-CD complex (B.E. = −30.461 kcal/mol) showed that the process of inclusion is spontaneous.

Side and top view of the a phenyl/β-CD and b Chlorophenyl/β-CD inclusion complex structure optimized by DFT (B3LYP)

Side and top view of the a phenyl/β-CD and b Chlorophenyl/β-CD inclusion complex structure optimized by DFT (M06L)
ROESY intensities studies
Intensity of ROESY peak directly depends on mixing time (τm) and cross relaxation rate (σ) between two spins (Eq. 1). The cross-relaxation rates between two spins depend on interproton distances (r−6), and the proportionality constant includes some physical constants with overall correlation time (τc) (Eq. 3). The physical constants included in the proportionality constant are same for all spin pairs in the given experiment. Therefore, the ratio of any two peak intensities of spin pairs can be considered equal to ratio of their interproton distances (Eq. 4). The use of these equations with their detailed description in determining interproton distances is reported in previous studies [8,9,10].
Therefore, determining interproton distances and their conversion into the intensities, can provide us useful information on stereochemical aspects and conformational details of an organic molecule. In a lot of examples this methology of converting interproton distances into intensities has been used to get highly atom accurate results [7].
In case of CTZ complexation with α-CD it was seen that only chlorophenyl ring of CTZ forms inclusion complex with α-CD. The three crosspeaks of which two appear below the diagonal and one above the diagonal, accurately represented the intermolecular interactions, one showing interaction between H6-9 aromatic protons and H3′ and H5′ cavity protons (ΣIH6-9/3′,5′), second and third peak having interaction of H6-9 with H3′(ΣIH6-9/3′)and H5′(ΣIH6-9/3′,5′) separately. From the experimental ROESY spectra it was found that ratio of intensities ΣIH6-9/3′:ΣIH6-9/5′:ΣIH6-9/3′,5′ is 0.90:1:2.07. Then the interproton distances between all aromatic protons of chlorophenyl ring of CTZ(H6-9) and CD cavity protons (H3′ and H5′) were measured, for the 3D models obtained from MM, MD and DFT (B3LYP, M06L). Then these distances were converted into intensities individually and then summed up to get ratio ΣIH6-9/3′:ΣIH6-9/5′:ΣIH6-9/3′,5′ accordingly. The calculated as well as experimental ROESY intensities for CTZ/α-CD complex are shown in Table 6.
In case of CTZ complexation with β-CD it was seen that both phenyl and chlorophenyl ring of CTZ forms inclusion complex with β-CD. The four ROEs two above the diagonal and two below the diagonal were seen useful in ROESY spectrum, one showing interaction between H6-9 aromatic protons and H3′ and H5′ cavity protons (ΣIH6-9/3′,5′), second peak showing interaction between H1-5 aromatic protons and H3′ and H5′ cavity protons (ΣIH1-5/3′,5′), second and third peak having interaction of H1-9 with H3′(ΣIH1-9/3′) and H5′(ΣIH1-9/3′,5′) separately. From the experimental ROESY spectra it was found that ratio of intensities ΣIH6-9/3′,5′: ΣIH1-5/3′,5′: ΣIH1-9/3′: ΣIH1-9/5′ is 0.54: 1: 0.64: 0.76. Then the interproton distances between all aromatic protons of both phenyl as well as chlorophenyl ring of CTZ (H1-9) and β-CD cavity protons (H3′ and H5′) were measured, for the 3D models obtained from MM, MD and DFT (B3LYP, M06L). Then these distances were converted into intensities individually and then summed up to get ratio ΣIH6-9/3′,5′: ΣIH1-5/3′,5′: ΣIH1-9/3′: ΣIH1-9/5′ accordingly. The calculated as well as experimental ROESY intensities for CTZ/β-CD complex are shown in Table 7.
Conclusion
We determine the atom accurate structures of the inclusion complexes of CTZ/α-CD and CTZ/β-CD. Chlorophenyl ring of CTZ forms an inclusion complex with α-CD, whereas both the aromatic rings of CTZ i.e. phenyl and chlorophenyl rings are involved in complexation with β-CD. All the inclusion complexes formed were found to have 1:1 stoichiometry. Minimum energy conformations from MM studies provided the inclusion mode and penetration depth of the guest. Least energy structures from the best MD runs whose calculated intensities matched the experimental ROESY intensities were considered the most accurate structures. ROESY peak intensities were calculated for the DFT (B3LYP) optimized structures and were in agreement with the experimental ROESY intensities. The calculated intensities for the structures obtained using DFT functional M06L, were not in good agreement with experimental ROESY intensities. Comparing intensities of structures obtained from our method and DFT optimized structures using B3LYP level of theory reveal the comparable accuracy of both methods.
References
Uhlenheuer, D.A., Petkau, K., Brunsveld, L.: Combining supramolecular chemistry with biology. Chem. Soc. Rev. 39, 2817–2826 (2010)
Ali, S.M., Muzaffar, S.: Quantitative ROESY analysis for unravelling structure of glafenine and β-cyclodextrin complex. J. Incl. Phenom. Macrocycl. Chem. 94, 95–102 (2015)
Schneider, H.J.: Mechanisms of molecular recognition: investigations of organic host-guest complexes. Angew. Chem. Int. Ed. Engl. 30, 1417–1436 (1991)
Lehmman, J., Kleinpeter, E.: 1H NMR spectroscopy as a probe of intermolecular interactions in β-cyclodextrin inclusion complexes. J. Incl. Phenom. Mol. Recognit. Chem. 10, 233–239 (1991)
Ali, S.M., Shamim, S.: Analysis of computational models of β-cyclodextrin complexes: structural studies of morniflumate hydrochloride and β-cyclodextrin complex in aqueous solution by quantitative ROESY analysis. J. Incl. Phenom. Macrocycl. Chem. 83, 19–26 (2015)
Ali, S.M., Upadhyay, S.K., Maheshwari, A.: NMR spectroscopic study of inclusion complexes of cetirizine dihydrochloride and β-cyclodextrin in solution. Spectroscopy 21, 177–182 (2007)
Muzaffar, S., Imtiaz, S., Ali, S.M.: Demonstrating accuracy of the proposed protocol for structure elucidation of cyclodextrin inclusion complexes by validation using DFT studies. J. Mol. Struct. 1277, 128419–128430 (2020)
Butts, C.P., Jones, C.R., Towers, E.C., Flynn, J.L., Appleby, L., Barron, N.J.: Interproton distance determinations by NOE—surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem. 9, 177–184 (2011)
Butts, C.P., Jones, C.R., Harvey, J.N.: High precision NOEs as a probe for low level conformers-a second conformation of strychnine. Chem. Commun. 47, 1193–1195 (2011)
Jones, C.R., Butts, C.P., Harvey, J.N.: Accuracy in determining interproton distances using nuclear overhauser effect data from a flexible molecule. Beilstein J. Org. Chem. 7, 145–150 (2011)
Chini, M.G., Jones, C.R., Zampella, A., Auria, M.V.D., Renga, B., Fiorucci, S., Butts, C.P., Bifulco, G.: Quantitative NMR-derived interproton distances combined with quantum mechanical calculations of 13C chemical shifts in the stereochemical determination of conicasterol F, a nuclear receptor ligand from Theonella swinhoei. J. Org. Chem. 77, 1489–1496 (2012)
Benghodbane, S., Khatmi, D.: Quantum chemical calculations based on ONIOM and the DFT methods in the inclusion complex: doxycycline/2-O-Me- β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 77, 231–240 (2013)
Katewongsa, P., Lertsuphotvanit, N., Phaechamud, T.: Cetirizine dihydrochloride, β-cyclodextrin inclusion complex by ethanol kneading for taste masking. Indian J. Pharm. 79, 758–767 (2017)
Stojanov, M., Wimmer, R., Larsen, K.L.: Study of the inclusion complexes formed between cetirizine and α-, β-, and γ-cyclodextrin and evaluation on their taste-masking properties. J. Pharm. Sci. 100, 3177–3185 (2011)
Allinger, N.L.: Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms 1,2. J. Am. Chem. Soc. 99, 8127–8134 (1977)
Klar, B., Hingerty, B., Saenger, W.: Topography of cyclodextrin inclusion complexes. XII.* Hydrogen bonding in the crystal structure of α-cyclodextrin hexahydrate: the use of a multicounter detector in neutron diffraction. Acta. Crystallogr. B36, 1154–1165 (1980)
Zabel, V., Saenger, W., Mason, S.A.: Neutron diffraction study of the hydrogen bonding in b-cyclodextrinundecahydrate at 120 K: from dynamic flip-flops to static homodromic chains. J. Am. Chem. Soc. 108, 3664–3673 (1986)
Neese, F.: The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012)
Paulino, P.H.S., de Sousa, S.M.R., Da Silva, H.C., De Almeida, W.B., Ferrari, J.L., Guimarães, L., Nascimento, C.S., Jr.: A theoretical investigation on the encapsulation process of mepivacaine into β-cyclodextrin. Chem. Phys. Lett. 740, 137060–137069 (2020)
Rafati, A.A., Hashemianzadeh, S.M., Nojini, Z.B., Safarpour, M.A.: Theoretical study of the inclusion complexes of α and β-cyclodextrins with decyltrimethylammonium bromide (DTAB) and tetradecyltrimethylammonium bromide (TTAB). J. Mol. Liq. 135, 153–157 (2007)
Pierre, M.D.L., Orlando, R., Mascho, L., Doll, K., Ugliengo, P., Dovesi, R.: Performance of six functionals (LDA, PBE, PBESOL, B3LYP, PBE0, and WC1LYP) in the simulation of vibrational and dielectric properties of crystalline compounds. The case of forsterite Mg2SiO4. J. Comput. Chem. 32, 1775–1784 (2011)
Grabowski, S.J.: Theoretical studies of strong hydrogen bonds. Annu. Reports Prog. Chem. Sect. C. 102, 131–165 (2006)
Acknowledgements
CTZ was obtained from Nebula Healthcare, India, and α-, β-CD were provided by Geertrui Haest, Cerestar Application center, Food & Pharma Specialities, France. Shah Imtiaz and Sughra Muzaffar are thankful to UGC, government of India for the financial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Imtiaz, S., Muzaffar, S. & Ali, S.M. Demonstrating accuracy of the already proposed protocol for structure elucidation of cyclodextrin inclusion complexes by validation using quantitative ROESY analysis. J Incl Phenom Macrocycl Chem 100, 71–87 (2021). https://doi.org/10.1007/s10847-021-01047-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-021-01047-9