
Abstract
In the twenty-first century, one of the central focus of polymer research in academia and industries is directed towards the design of environmentally-benign materials produced from reagents that have minimal deleterious effects on our environment. The aliphatic polyester PLA is one such example. Due to its biodegradable, biorenewable and biocompatible nature, PLA finds diverse applications, especially in the biomedical field. PLA is exclusively synthesized by the ring-opening polymerization of lactide (cyclic dimer of lactic acid) in the presence of a catalyst. The macrocycles and macrocyclic metal moieties can act as effective catalysts for the polymerization resulting in the formation of PLA with controlled tacticity and predetermined molecular weight. This review reports metal-based catalytic systems supported by porphyrin, calixarene and bispyrrolidine- salan as ancillary ligand and metal-free organocatalyst sparteine for the ROP of LA. The variation in catalytic activity, tacticity of PLA, and PLA's molecular weight distribution by substitutional changes in the catalyst framework have been discussed in detail.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Background
Polymers find extensive use in our daily life. The polymers can either be naturally occurring like protein, starch, DNA, or synthetic which include polyethene, polyvinyl chloride, polyesters, and many others. Among the synthetic polymers known to date, those derived from petrochemical-based sources have innumerable advantages [1]. Nevertheless, they have two significant limitations, namely utilization of non-renewable petroleum sources for production and the factor of their non-biodegradability, which restricts their plausible use in the long run [2]. Thus, there has been a tremendous increase in research, focusing on the manufacture of polymers derived from renewable sources.
The synthetically derived biodegradable polymers mainly consist of polyesters, polyamides, polycarbonates, polyanhydrides, etc. [3]. Among the different synthetic biodegradable polymers obtained from renewable sources known to date, aliphatic polyesters (PLA, PGA, PLGA, PCL) are of immense importance. Due to its biodegradable and biocompatible nature, aliphatic polyesters find important applications in the biomedical and food packaging industry [4,5,6,7].
PLA has received immense importance as a suitable substitute for polymers derived from petrochemical-based sources as its monomer (lactic acid) is procured from annually renewable sources like corn and beetroot [8]. In addition, PLA undergoes hydrolysis under the physiological condition to give non-toxic product lactic acid, which eliminates from the environment as carbon dioxide and water as a result of microbial action [9]. The synthesis of PLA is done through either direct polycondensation of lactic acid or ROP of lactide [10]. The polycondensation pathway is less significant as water liberates continuously, which is difficult to remove, and severe reaction conditions are required to obtain a polymer with high molecular weight [11]. Thus, the ROP of lactide has drawn considerable attention as it allows better control over molecular parameters (polydispersity index, molecular weight) under mild reaction conditions in comparison to the polycondensation pathway [12].
Metal containing complexes [13,14,15,16,17] and organocatalysts [18, 19] generally catalyse the ROP of LA. The metal-based catalytic system consists of a metal center supported by an ancillary ligand and sometimes an initiating group attached to the metal center. The electronic and steric environment around the metal center governed by the ancillary ligand plays a crucial role in regulating molecular parameters of the polymer and has consequent effects on its stereoregularity [20,21,22].
The first macrocyclic polyether was developed in 1967 by Charles Pedersen [23]. He named these macrocycles as “crown ethers” as its molecular array is shaped like a crown. The discovery of Pederson’s “crown ether” opens a variety of research scope in macrocyclic chemistry. Host–guest chemistry [24] and supramolecular chemistry [25] associated with crown ethers has become a prominent area of research. Living creatures regularly produce tetrapyrrolic macrocyclic systems in their body, either as a metal free form (e.g., pheophorbide) or, as its metal complexes (e.g., chlorophyll, coenzyme B12, heme). These are named, “the pigments of life” as they execute a variety of fundamental biological functions necessary for living [26]. Calixarenes are an important class of compounds in the macrocyclic family and have been used for applications in the treatment of cancer due to its special molecular framework [27]. Calixarenes have large available pores and cavities and may be used for storage of gases [28]. The advantage of these systems is they remain stable and flexible for larger calix[n]arenes (n > 4) and can accommodate large metals along with its bulky substituents [29]. The porphyrin system with its different metal analogues and metallocalix[n]arenes show diverse reactivity in ROP reactions due to their rigid framework. The half-porphyrin analogues and their saturated amine complexes constitute the diverse category of metal salen and salan compounds and they have exhibited their profound role in metal catalysed ROP reactions in the recent years [30,31,32,33,34,35,36,37]. However, salan analogue with the bispyrrolidine system of ligand give us a rigid framework analogous to salen and other macrocycles, having diverse reactivity with metal synthons and producing complexes which show better control over the ROP of LA [38]. Hence, it is pertinent to include the chemistry and ROP results of these complexes within the scope of this review.
The organocatalysts are the metal-free catalytic systems that follow an ionic (cationic or anionic) or an enzyme-catalysed pathway for ROP [39, 40]. Macrocycles can act as a prominent auxiliary ligand in metal complexes and can even act as a suitable component of metal-free catalyst for the ROP of lactide. The metal-free organocatalysts are now being used vigorously for the controlled polymerization of LA [41,42,43,44,45,46]. Sparteine is a macrocyclic Lupin alkaloid [47], which is being used as a prominent organocatalyst towards the ROP of LA [48]. In this review, we shall elaborate the behaviour of sparteine towards the ROP of LA.
In the recent years, modification of PLA with certain additives with the intention of enhanced properties required for futuristic applications has been actively reported. Different methods for the synthesis of lactic acid oligomers modified with macrocyclic fragments like porphyrin, cyclodextrin and cyclophane are reported that has led to enhanced performance of oligolactide containing sensors and biosensors [49]. The synthesis of oligolactide and PLA functionalized with p–t-butylthiacalix[4]arenes is recently reported to exist in three conformations namely cone, partial cone, 1,3-alternate which were synthesized by ROP [50].
Scope
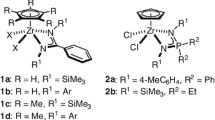
The present review shall focus on the macrocyclic systems namely porphyrin, calixarene, and bispyrrolidine salan as ancillary ligands (Fig. 1) in metal complexes used for ROP. The metal complexes of these ligands have been used towards the ROP of LA. In addition, the application of sparteine (a chiral amine) (Fig. 1) towards the ROP of LA as a metal-free catalyst shall be described. The role of the metal center, the change in the steric and electronic environment of the parent macrocycle by suitable substitution in the ligand backbone, in controlling molecular parameters of the polymer during ROP shall be highlighted.

Metal complexes of macrocycles in this review
Mechanistic pathway for rop of lactide
Coordination-insertion mechanism
The ROP of LA catalysed by metal complexes mostly takes place by the coordination-insertion mechanism. In the first step, the monomer coordinates to the Lewis acidic metal center of the metal complex MLX (M = metal, L = ligand, X = initiating group) followed by insertion of monomer in the M − X bond through a nucleophilic attack by X on carbonyl carbon. Next acyl − oxygen bond cleavage results in the ring-opening step and polymerization continues until hydrolysis occurs and results in chain termination (Scheme1) [51].

Coordination-insertion mechanism for ROP of LA
Macrocycles as ancillary ligand in metal complex in rop of lactide
Porphyrin
Generally, porphyrins are used as substituted porphins. Porphin is a macrocyclic moiety that has a planar structure with four pyrrole-like rings connected by methine bridge. Porphyrin is aromatic due to conjugation of the18π electrons [52].
In 1987, Inoue and co-workers for the first-time reported novel aluminium tetraphenyl porphyrin (TPPAl) complexes that act as initiators for the ROP of D-LA [53]. Four different Al-porphyrin complexes (Fig. 2) were produced by different synthetic pathways. The complex (TPP)AlPPO (2a) was prepared by the polymerization of 1,2-epoxypropane with (TPP)AlCl in ratio (20:1). Again, (TPP)AlOCH3 (2b) was prepared by the reaction of (TPP)AlCl with CH3OH under nitrogen atmosphere. The limiting synthons namely, (TPP)AlEt (2c) and (TPP)AlCl (2d) were synthesized by the reaction of (TPP)H2 with Et3Al and Et2AlCl respectively in dichloromethane (CH2Cl2) under nitrogen atmosphere at room temperature (Scheme 2). These compounds were characterized by 1H NMR spectroscopy. These complexes were screened for the polymerization of D-LA. PLA with low PDI values were obtained with (2a, 2b, 2c) and the yield of the polymer was high only at high temperature and prolonged reaction time. Among these complexes, the best result was found for (2a) [[D- LA]/ (TPP)AlPPO] = 100], with 94% conversion found in 96 h, but (2d) does not show any catalytic activity due to the poor nucleophilicity of chloride as an initiating group. The mechanism was found from 1H NMR analysis. A reaction mixture containing (TPP)AlOCH3 (2b) and D-LA (1:5 molar ratio) at room temperature was subjected to 1H NMR (CDCl3) analysis after 1 h of runtime. The spectrum shows signals at [ẟ = 9.1 (pyrrole-H), ẟ = 8.2 and 7.8 (aromatic protons of phenyl ring)] and ẟ = 1.65 and 4.95 due to methyl and methine hydrogens of D-LA. Finally, the appearance of three new signals clarifies the mechanism [two doublet a (ẟ = − 1.56) and a′ (ẟ = 0.97), two quartet b (ẟ = − 1.95) and b′(ẟ = 3.95) and a singlet c (ẟ = 3.29)] (Scheme 3). The signal at ẟ = − 1.3 ppm corresponding to the Al-OCH3 group disappeared completely. Thus, the reaction proceeds via acyl − oxygen bond cleavage, resulting in (porphinato)-aluminium alkoxide intermediate that acts as the chain propagating species (Scheme 3).

Al-porphyrin complexes

Synthesis of aluminium complexes

Reaction between (TPP)AlOCH3 and D-LA
Gao and Duan's group synthesized three aluminium complexes bearing porphyrin derivatives as an ancillary ligand [54] (Scheme 4). For the synthesis of these complexes, a mixture of the respective ligand and trimethylaluminium were mixed in equimolar ratio in toluene at 40 °C and stirred for 12 h under an argon atmosphere. The three complexes differed from each other by different substituents at the phenyl/pyrrole rings of the parent porphyrin. The three complexes were characterised by NMR studies and elemental analysis. The 1H NMR (CDCl3) spectrum of the ligands showed characteristic peak for the − NH proton (marked as 1, Scheme 4) in the region ẟ = 2.74 to 3.20 ppm. However, upon the addition of the aluminium alkyl, the disappearance of signal for NH proton in the aluminium complexes and the appearance of a new signal for protons of the methyl group attached to aluminium [ẟ = − 6.82 ppm(3a); ẟ = − 6.90 ppm(3b) and ẟ = − 6.92 ppm(3c)] provided evidence for the formation of these new complexes. These complexes were evaluated towards the ROP of LA in the presence of different solvents namely THF and toluene in the presence isopropanol as co-initiator at 70 °C.

Synthetic route for aluminium porphyrin complexes
Under identical reaction conditions, complex (4a) with least substituents on the ligand backbone showed the highest activity (% conversion = 93) towards ROP. The microstructural analyses of the polymer obtained by the three catalysts indicate that polymers were slightly isotactically enriched in all the cases. The corresponding Pm values were [4a:(Pm = 0.53), 4b:(Pm = 0.59) and 4c:(Pm = 0.65)]. Under the same temperature and [monomer]/[catalyst] ratio, complex (4c) showed the highest degree of stereoselectivity in toluene. The experimental results revealed that steric bulk of porphyrin ligand had a strong influence on the tacticity of the PLA. With the pyrrolyl moiety being closer to the metal center, any substituent on the pyrrolyl ring has a much greater influence on the tacticity of the PLA as compared to the substituents on the phenyl ring. For complex (4c), lowering the temperature from 70 °C to 40 °C and changing the solvent from toluene to THF increased Pm value from 0.65 to 0.68 indicating that the tacticity of the polymer is influenced by the reaction temperature. The kinetics study of the polymerization reaction with complex (4c) along with isopropanol as the co-initiator at 70 °C in toluene revealed for [[LA]/[Al complex] = 100:1], the molecular weight of polymer increased linearly with increase in monomer conversion rate and PDI value varied between 1.08 to 1.11, indicating living nature of the polymerization. The 1H NMR studies of the polymer obtained by using complex (4c) revealed the polymer to be end capped by a hydroxyl group and an isopropyl ester group. The 1H NMR (CDCl3) signals [Hf (ẟ = 1.24 ppm):Hd (ẟ = 4.35 ppm) = 6:1] (Fig. 3) gives evidence that the ROP proceeds via coordination-insertion mechanism.

PLA with hydroxyl end group and isopropyl ester end group
Chisholm reported the action of TPPCr(III) (Scheme 5) towards the ROP of L-LA and rac-LA [55] in the presence of PO. The complex (TPP)CrCl was prepared by the earlier reported procedure by reacting excess CrCl2 with TPPH2 in dimethylformamide (DMF) under reflux conditions followed by an aqueous work-up [56]. Anhydrous CrCl2 was added to a solution of TPPH2 in DMF in four lots under reflux conditions. The reaction was monitored by recording the visible spectrum of the reaction mixture after the addition of each lot. The absence of signal of free porphyrin in the visible spectrum of the reaction mixture indicated completion of the reaction. The group had reported earlier that TPPCr(III)Cl is a better catalyst than structurally similar Al(III) analogues for the polymerization of propylene oxide [57].

Synthesis of TPPCrCl
A polymerization trial was done at 0 °C using TPPCrCl, PPN+Cl−, LA and PO leading to the formation of PLA with − OCHMeCH2Cl as an end group as understood by mass spectrometry. However, the reaction mixture contained a small fraction of PLA containing the hydrolyzed end group namely − OCHMeCH2OH. Under similar conditions, rac-LA yielded almost isotactic PLA as confirmed from the results of homonuclear decoupling 1H NMR and powder X-ray diffraction studies. A careful analysis of this phenomena revealed that towards the end of the polymerization reaction, a new six-member ring product is formed from PO (3,6-dimethyl-1,4-dioxan-2-one) as a result of a backbiting reaction (Scheme 6). This product does not polymerize at 0 °C.

Plausible pathway for formation of six membered lactone by backbiting mechanism
Authentic samples of this product were obtained by reacting L-ethyl lactate and allyl bromide leading to four stereoisomers (Fig. 4) which were systematically characterized through 1H and 13C NMR spectroscopy and gas chromatography using columns with achiral and chiral stationary phases.
At 0 °C, the reaction between L-LA and (S)-( −)- or (R)-( +)-PO yielded (4a) or (4b) as the major product. This shows that the stereochemistry of the PO employed is retained in the product.
The results are different at 60 °C. The varying percentages of 3,6-dimethyl-1,4-dioxan-2-one stereoisomers (Fig. 4) received with different combinations of LA and PO isomers at 60 °C is depicted in Scheme 7.

Stereoisomers of 3,6-dimethyl-1,4-dioxan-2-one

Product profile at 60 °C under different combinations of LA and PO
This shows that at higher temperatures, the formation of two diastereomers is significant unlike what is seen from the results at 0 °C (Scheme 8)

Product profile at 0 °C under different combinations of LA and PO
The same research group reported a new synthetic pathway for the polymerization of rac-LA at room temperature using an aluminium catalyst system (Fig. 5) [58] (Scheme 9). The aluminium complexes were characterised by 1H NMR studies. For all the five complexes, 1H NMR showed three distinct proton signals in the aromatic region.

Aluminium porphyrin complexes

Synthesis of aluminium porphyrin complexes
The tetraphenylporphyrinAl(III) salts TPPAlX (X = CH3, Cl, OEt, OiPr, OCH2CHMeCl) in the presence of PPN+Cl−, and propylene oxide polymerize rac-LA to give isotactic PLA and with time it gives cyclic polymer (PO)nPLA. The reaction of rac-LA with TPPAlCl/PPN+Cl− in rac-PO at room temperature gave isotactic PLA with MW = 3500 Da and PDI 1.08. The Al(III) center being a Lewis acid coordinates to the oxygen atom of propylene oxide, thus facilitating attack by Cl− of PPN+Cl−, resulting in the ring-opening of propylene oxide leading to the formation of chiral X(TPP)AlClCH2*CHMeO−. The chiral alkoxide X(TPP)AlClCH2CHMeOC(O)CHMeC(O)CHMeO− generated by reaction of X(TPP)AlClCH2CHMeO− and rac-LA further react with unreacted rac-LA to give PLA (Scheme 10). The MALDI-TOF spectrum of the PLA showed the appearance of PLA with terminal OCHMeCH2Cl giving evidence for the formation of AlOCHMeCH2Cl. The possibility of anionic polymerization is eliminated as the reaction involving rac-LA and PPN+Cl−, does not give PLA. There is no PLA formation when TPPAlOR (OR = OEt, OiPr, OCHMeCH2Cl) along with PPN+Cl− and rac-LA in THF at room temperature is employed for the polymerization, indicating the alkoxides do not act as initiator for the ROP of LA at room temperature. When the reaction is carried in the presence of PPN+NO3−, the product is not formed, indicating that a suitable nucleophile is required for the ring-opening of PO. The formation of cyclic polymer can occur due to the ring-opening of epoxide present as the solvent by an alkoxide ClCH2CHMeO(LA1/2)nC(O)CHMeO− and the new alkoxide ClCH2CHMeO(LA1/2)nCH2CHMeO− can back-bite its own C − Cl group.

Plausible initiation step for the ROP of LA via ring-opening of PO with PPN+Cl−
Inspired by the work of Chisholm et al. on the ROP of LA at room temperature, Phomphrai and co-workers investigated different metal (III) centers with tetraphenylporphyrin and cyclohexyl-salen (Cy-salen) as ligands for the polymerization of rac-LA [59] (Fig. 6) (Scheme 11). The authors were inspired to use a different epoxide namely CHO and a combination of different metal centers to study the effects on the polymerization of LA.

Metal complexes with TPP and Cy-salen ligand

Synthesis of metal-porphyrin and metal-salen complexes
The effect of the metal center, ligand environment and co-catalyst were studied in detail. CHO being more sterically hindered than PO was used for the polymerization study (Scheme 12). The order of reactivity for TPP supported metal (III) catalyst was Cr(III) > Al(III) > In(III) > Co(III) (% conversion: TPPCrCl = 95% in 30 min, TPPAlCl = 84% in 1 h and TPPInCl = 94% in 11.5 h and TPPCoMe = 22% in 25 h) which is accordance with the Lewis acid character of the metal center. For Cy-salen ligated metal(III) center, the order of reactivity was In(III) > Cr(III) > Al(III) > Co(III) [% conversion (Cy-salen)InCl = 95% in 3 h, (Cy-salen)CrCl > 99% in 3 h and (Cy-salen)AlCl: > 99% in 21 h] which is attributed to the large size of In(III) that perfectly fits in the flexible pocket of the ligand and can easily be accessed for CHO coordination. For the three initiators namely PPN+Cl−, DMAP, pyridine chosen for the polymerization reaction, the ionic PPN+Cl− showed the highest activity, followed by neutral DMAP and pyridine which is in accordance with nucleophilic character of the co-catalyst. The conversion percentage of monomer for complex TPPAlCl for different co-catalysts investigated at 30 °C was PPN+Cl− = 84% in 1 h, DMAP = 55% in 1 h and pyridine = 80% in 20 h respectively. Increasing the amount of co-catalyst decreased the reaction time but the PDI value of the polymer increased. For aluminium complexes TPPAlX (X = Cl, Me, OMe, OCHMeCOOEt) though X group is not an initiator but its electronic contribution affects catalyst activity. The highest catalytic activity was observed for TPPAlOCHMeCOOEt, (% conversion > 99% in 30 min for [monomer]/[catalyst] = 100:1 at 30 °C) indicating that a higher electrophilic ligand favours the increase in catalytic activity. The polymerisation of rac-LA using rac-SO showed very little activity.

Plausible initiation step for the ROP of LA via ring opening of CHO with PPN+Cl− and intramolecular backbiting mechanism
The mechanism can be explained by MALDI-TOF spectrometry. The major An series in MALDI-TOF spectrum signifies that ¯OCH(CH2)4CHCl [i.e. (CHO)Cl alkoxide] act as the initiating alkoxide which was formed by ring opening of CHO by Cl−, propagating the PLA chains (Scheme 12). A minor series Bn was observed corresponding to the formation of a cyclic species as indicated in Scheme 12.
Duan and Gao research group prepared three aluminium porphyrin complexes (Scheme 13) and used them for the polymerization of L-LA in the presence of copper-porphyrin as co-catalyst [60] (Scheme14). The complexes were characterized by 1H NMR spectroscopy. The polymerizations were done in toluene at 70 °C. The rate of polymerization increases with the introduction of the electron-withdrawing substituent in the phenyl moiety as this results in weakening of the Al-alkoxide bond. This is evident from % conversion values (13a = 80.2%, 13b = 74.3% and 13c = 93.0%). There is a significant decrease in monomer conversion with an increase in steric bulk of the group attached to the phenyl ring. The mechanistic aspects were understood through the end group analysis of the oligomer obtained by the polymerisation of L-LA at low monomer to initiator ratio ([monomer]:[Al-complex 13b]:[Cu-complex] = 16:4:1). The 1H NMR (CDCl3) spectra of oligomer showed ẟ = 4.34 ppm for the methine proton attached to hydroxyl (marked as a) end and ẟ = 8.67 ppm for hydrogen attached to the pyrrole ring (marked as b). Hence, the oligomer is end-capped by hydroxyl group at one end and porphyrin benzyl ester at the other end (Fig. 7). There was initial formation of a copper core with four side chains of porphyrin benzyl aluminium species. The aluminium alkoxide acts as an initiator for the polymerization leading to the formation of functionalized PLA (Scheme 15).

Synthesis of aluminium porphyrin complex

Synthesis of copper co-catalyst

Poly (L-LA) oligomer

Mechanism for ROP of LA with aluminium porphyrin complex and Cu-complex
Calixarene
Calixarenes are macrocycles prepared by base-catalysed condensation of formaldehyde with suitable para-substituted phenols. In Calix[n] arene ‘n’ denotes the number of phenolic units present. The ortho positions of phenyl ring are connected by methylene bridges [61].
In 2005, Vigalok and co-workers prepared bimetallic alkylzinc (alkyl = ethyl and methyl) calixarene complexes to evaluate single-site catalytic mechanism in the bimetallic Zn system (Scheme 16) [62]. The bimetallic ethylzinc complex was prepared by overnight stirring of the reaction mixture formed by the reaction of 1,3-di-O-n-propyl-t-butyl substituted calixarene in toluene with Et2Zn. The 1H NMR studies revealed that the two ethyl groups showed two different signals which gave evidence that the two zinc atoms were not in the same electronic environment. The methylzinc complexes were prepared by the overnight stirring of di-n-propyl ether solution of t-butyl substituted calixarene in toluene and Me2Zn. Again, the methyl group attached to the zinc center gave two distinct signals in the 1H NMR which supported the fact that the two zinc atoms are not equivalent. The 1H NMR observations indicate that one zinc atom is entrapped in the calixarene cavity, and other zinc atom lies outside the cavity. The reactivity of these Zn(II) centers towards ROP were found to be different.

Preparation of zinc calixarene complexes
The ROP of L-LA was carried out at 60 °C in toluene with zinc calixarene as the catalyst. The initiation starts with the alkylzinc group located outside the calixarene cavity, and the propagation continues with the zinc alkoxide complex formed after the first insertion (Scheme 17). The initiation step is the rate-limiting step of the reaction as the alkylzinc group is a poor nucleophile. The Zn alkoxide formed after the first insertion being a much better nucleophile than the alkylzinc component resulted in polymer with good molecular weight even at low [monomer]/[catalyst] ratio as chain propagation continues with the zinc alkoxide complex generated in situ. The 1H NMR of the methylzinc complex gave two signals at ẟ = 0.01 and –2.32 ppm corresponding to hydrogen atom of methylzinc moiety located outside and inside calixarene moiety respectively. The methylzinc group inside the calixarene cavity showed more upfield shift as it lies in the shielding region of four aromatic rings. The initiation does not involve the zinc fragment inside the calixarene cavity as evident from the 1H spectrum of the pure polymer isolated after quenching that showed a signal at ẟ = − 2.18 corresponding to methyl group attached to zinc located inside the cavity. Replacing the ethylzinc moiety with methylzinc as an initiating group improved the catalytic activity and the PDI value of polymer decreased from 1.45 to 1.06. The two alkylzinc complexes (16a) and (16b) were further reacted with Me2Zn and Et2Zn respectively (Scheme 18). The catalytic activity of complex (18a) and (18b) resembled (16b) and (16a) respectively. Further, the polymerization reactions carried out with 1: 1.5 mixture of (16a) and (18b) gave only one product as indicated by the single peak in the GPC trace that gave evidence that only the alkylzinc moiety located outside calixarene cavity participates in the initiation process.

Polymerization of L-LA

Synthesis of bimetallic alkylzinc with different substitution
Frediani and research group synthesized chlorotitanium calix[4]arene complex 5,17-bis-t-butyl-11,23-dinitro-25,27-dipropyloxy-26,28-dioxo-calix[4]arene titanium dichloride (Scheme 19) and investigated its catalytic activity towards the polymerization of L-LA and rac-LA in the absence of solvent [63]. For the synthesis of the titanium complex, a solution of TiCl4 in dichloromethane was added dropwise to the solution of 5,17-bis-t-butyl-25,27-dihydroxy-11,23-dinitro-26,28-dipropyloxy-calix[4]arene in toluene at room temperature, and the resulting red solution was heated at 60 °C for 48 h. The product was precipitated by the addition of hexane. The product was characterized using 1H NMR and X-ray diffraction studies. The 1H NMR studies revealed the Ti-complex to have C2v symmetry. The single-crystal X-ray diffraction study of the Ti complex revealed the complex to be triclinic and it crystallises with 1.5 molecules of CH2Cl2. In the complex, the Ti atom is in an octahedral coordination environment consisting of two chlorine atoms and four oxygen atoms. The two propylated oxygen occupy a trans position. For the polymerization of L-LA, varying [monomer]/[catalyst] ratios from 196 to 1900 were used. The time of polymerization was kept constant. An increase in activity of the polymerization by eight times and decrease in PDI value of PLA from 1.4 to 1.2 was observed. There was a significant difference between experimental and theoretical molecular weight due to intramolecular transesterification reactions. Mechanistically, the complex acts as a dual-site catalyst resulting in the insertion of two monomer molecules into each Ti − Cl bond that leads to the growth of two separate polymeric chains at the same Ti center (Scheme 20). The MALDI-TOF analysis of PLLA showed peaks corresponding to half the molecular weight of the monomer that supports intramolecular transesterification arising due to dual-chain growth from a single Ti atom (Scheme 21). The absence of peak corresponding to cyclic PLA in the MALDI-TOF spectrum further supports dual chain growth from single titanium atom as cyclic PLA must arise due to intramolecular transesterification of a single chain from the Ti center. Addition of butanol increased the rate of polymerization and decreased the PDI value of PLA to 1.1. This is attributed to the formation of Ti-dialkoxyde, which catalyses the polymerization reaction at a faster rate (Scheme 22). The microstructural analysis of the polymer indicated the formation of an isotactic PLA. The polymerisation studies for rac-LA showed the same activation rate as for L-LA for a molar ratio between LA and the catalyst of 463.

Synthesis of Ti complex

Two-site ROP

Intramolecular transesterification

Transfer reaction by butanol
Frediani et al. synthesised modified version of Ti calix[4]arene complex (Scheme 23) to study ROP of rac-LA under thermal activation and microwave irradiation [64]. The Ti (IV) complex is prepared by reacting 25,27-dipropyloxy-26,28-dihydroxy-calix[4]arene with 1 M TiCl4 in toluene at 60 °C (Scheme 23). The 1H and 13C NMR spectra of the Ti-complex gave evidence of C2v symmetry. The X-ray crystal structure further revealed an octahedral coordination environment around the Ti(IV) center with the Ti being coordinated to four oxygen atoms and two chlorine atoms.

Preparation of Ti complex
Under microwave irradiation, polymerization rate increased in comparison to the thermal method. For [monomer]/[catalyst] = 200, the conversion reached 96% in 3 h under thermal condition in comparison to 88% conversion achieved in just 80 min under microwave irradiation. The PLA obtained under thermal activation gave slightly better PDI value (PDI = 1.2) than microwave irradiation (PDI = 1.3). The microstructural analysis of the polymer shows the PLA to be partially isotactic stereoblock and similar microstructures were obtained even by varying the [monomer]/ [catalyst] ratio.
A heterobimetallic complex [Li(THF)Mg(nBu)L] containing lithium and magnesium supported by the ligand 1,3-dipropoxy-p–t-butylcalix[4]arene (LH2) and a mononuclear magnesium complex having tripropoxy-p–t-butylcalix[4]arene as the ligand backbone were subsequently reported by Redshaw (Scheme 24) [65]. The single-crystal X-ray structure of complex (24a) showed that the calixarene moiety adopts an elliptical conformation, where in the Li atom along with one THF molecule resides in the cavity of the calixarene. The THF is labile and no signal corresponding to it appears in the 1H NMR spectrum. The magnesium atom adopts distorted trigonal bipyramidal geometry with the magnesium atom coordinated to four oxygen atoms and the fifth coordination is satisfied by the n-butyl group. The oxygen atom of THF, Li, Mg and the carbon atom of the n-butyl group are linear. The solid-state structure of the complex (24b) revealed that the complex crystallizes along with a molecule of pentane. The Mg atom adopts trigonal bipyramidal geometry with the Mg atom coordinated to four oxygen atoms of calixarene with the fifth site occupied by the n-butyl group. Both the complexes are active for the ROP of rac-LA. The heterobimetallic complex, in combination with co-initiator MeOH in CH2Cl2, gave 94% conversion in 2 h producing PLA with PDI = 1.19. In the absence of alcohol co-initiator, (24a) is inactive for the ROP of rac-LA and other co-initiators such as isopropanol, t-butanol and benzyl alcohol resulted in low activity. The mononuclear Mg complex is very active and provided PLA with narrow dispersity index. However, (24b) showed the highest activity in toluene and THF in comparison to CH2Cl2. In contrast to (24a), addition of isopropanol, t-butanol and benzyl alcohol gave better activity in comparison to methanol. The use of methanol in THF resulted in lower activity in comparison to the situation where no alcohol was used. The results indicate methanol deactivates the catalyst to a certain extent. The complex (24b) shows 92% conversion in just 3 min in the presence of benzyl alcohol as co-initiator and THF as the solvent. Interestingly, this mononuclear complex behaves differently in different solvents. In THF it shows an immortal polymerization behaviour producing PLA with heterotactic bias (Pr = 0.85), but in toluene, the PLA obtained had an isotactic bias (Pr = 0.30–0.36). On the other hand, both complexes (mononuclear and heterobimetallic) gave atactic PLA in CH2Cl2.

Synthesis of heterobimetallic Li and Mg complex (24a) and mononuclear Mg complex (24b)
Bispyrrolidine salan
The bispyrrolidine salan system is well known [66] and is comparable to half porphyrin and salen systems. The bispyrrolidine salan has two sp3 nitrogen atoms where as a conventional porphyrin has four sp2 nitrogen atoms. The bispyrrolidine salan containing catalytic systems have diverse and fascinating reactivity features towards ROP reactions [67].
Jones reported three Zr(IV) complexes with an analogue of salan, incorporating enantiopure [R,R (1) and S,S (2)] and meso(3) variants of 2,2′-bispyrrolidine (Fig. 8) [68]. The three complexes were characterized by single-crystal X-ray diffraction. The X-ray diffraction studies of the complexes revealed that enantiopure 2,2′-bispyrrolidinesalanZr(IV)(OiPr)2were present as ∆- and Ʌ- isomers respectively in the solid-state. In the complex, the enantiopure ligands were bound to the Zr(IV) center in a "fac-fac" fashion since the hydrogen atoms of the C − C bond between two five-membered rings of 2,2′-bispyrrolidine salan are antiperiplanar to each other. In the meso-2,2′-bispyrrolidinesalanZr(IV)(OiPr)2 complex, the ligand was bound to the Zr(IV) center in a "fac-mer" fashion where the hydrogen atoms, attached to the C − C bond between five-membered rings of bispyrrolidine salan are syn to each other (Fig. 9). The complex with the meso ligand had both ∆ and Ʌ isomers in the solid-state. However, in the solution phase it showed fluxionality where both ∆-isomer and Ʌ-isomers were in equilibrium and inter-convertible at room temperature. On the other hand, for the metal complexes formed with enantiopure ligand, the ligand backbone was locked in the solution phase similar to that in the solid-state. Hence, only one isomeric form (∆ or Ʌ) was present in the solid phase as well as in solution phase. These observations were confirmed by NOESY/EXSY measurements where the complex with metal atom bound to the meso ligand showed some exchange peaks under ambient temperature. These peaks were absent for complexes with enantiopure ligands attached to the metal center.

Different 2,2′-bispyrrolidine salan systems

fac-fac and fac-mer binding in 2,2′-bispyrrolidinesalanZr(IV) isopropoxide
During polymerization with rac-LA, meso-2,2′-bispyrrolidinesalan Zr(IV) complex produced a highly isotactic block PLA (Pm = 0.86) with 85% conversion in 8 h at room temperature. The block nature of the polymer can be explained from the proton decoupled NMR spectrum where the peak intensity of 'sii', 'iis' and 'isi' tetrad is significantly higher than 'sis' tetrad. However, the complexes bound to enantiopure ligands were inactive for ROP but at 70 °C in toluene, they produced PLA with strong isotactic bias (Pm = 0.80, 0.80) after 8 h with 54% and 60% conversion respectively. From the kinetics study, it was observed that Ʌ-Zr(S,S)(OiPr)2 specifically polymerizes L-LA, which is an indication that the polymerization goes with enantiomorphic site controlled mechanism [69]. The mechanism for controlled polymerization of rac-LA by meso-2,2′-bispyrrolidinesalanZr(IV)(OiPr)2 also follows the enantiomorphic site control pathway since ∆-and Ʌ- isomer of Zr(meso)(OiPr)2, preferentially coordinate to the D-LA and L-LA respectively (Scheme 25).

Interconverting isomers produce stereoblock isotactic PLA
Subsequently, the same research group reported Ti(IV), Hf(IV) and Al(III) isopropoxide complexes with isomeric [(R,R) (1), (S,S) (2)] and [(meso)(3)] 2,2′-bispyrrolidine salan ligand [70]. In the Hf(IV) complexes, the enantiopure and meso ligand variants bind in α-cis and β-cis geometry respectively. In the solution phase, the complexes bound to the enantiopure ligand were locked. However, ligand exchange happens in case of the complex from the meso ligand due to the fluxionality in the molecule, as seen previously with the Zr(IV) analogues [63]. For ROP of rac-LA, fluxional meso(3)Hf(IV) complex formed stereoblock isotactic PLA (Pm = 0.84) (similar to Scheme 25) with 87% conversion in 4 h at 50 °C in toluene. On the other hand, the Ti(IV)mesobispyrrolidinesalan complex formed atactic PLA with only 48% conversion in 24 h at 130 °C under solvent free melt condition.
The methyl Al(III) complexes with enantiopure bispyrrolidine salan system, had a trigonal bipyramidal geometry [τ = 0.97 for the RR system where, τ is geometry index or structural parameter [71, 72] but its isopropoxide analogue had pseudo trigonal bipyramidal structure (τ = 0.70 for the RR system). The aluminium isopropoxide complexes where the metal center is attached to enantiopure ligands were found to be fluxional in the NMR time scale and produced atactic PLA in both melt and solution phases. On the other hand, the aluminium complex of the meso bispyrrolidine salan system (Fig. 10), had a pseudo trigonal bipyramidal geometry (τ = 0.64) and was found locked both in solution and solid phases. It produced heterotactic PLA (Pr = 0.87) in toluene at 70 °C (Scheme 26).

mesobispyrrolidinesalanaluminiumisopropoxide complex

Different bispyrrolidine salan complexes and their lactide polymerization tacticity
Jones and co-workers synthesized Zn(II), Mg(II) and Li(I) complexes [73] with the same variant of bispyrrolidine salan ligands reported earlier (Fig. 8) [68]. They also synthesized a cubic Li3Mg1O4 core structured complex with the meso bispyrrolidine salan. This was prepared by a 1:1:1 reaction of meso bispyrrolidine salan, nBuLi and nBu2Mg. The Li4(3)2(THF) complex was prepared using nBuLi as the Li precursor and the meso variant of the ligand. The complex formed a Li4O4 cube where all the Li atoms were coordinated to three oxygen atoms. Among the four lithium atoms present in the cubic core, two planar lithium atoms were connected further to two nitrogen atoms. The third lithium atom is coordinated to THF and the fourth lithium showed weak η1-Ph interaction with the phenyl ring. The lithium complexes with enantiopure ligands could not be isolated due to high solubility in common organic solvents. The reaction between the meso ligand and ZnMe2 gave a metal complex Zn4(3)2(Me)2(OMe)2 (Fig. 11) where two zinc atoms are four coordinated and remaining two zinc atoms are five coordinated. The methoxy moiety came from the insertion of adventitious oxygen present in the solvent into Zn-C bond. The reaction of enantiopure bispyrrolidine salan derivative with MgnBu2 gave phenoxide bridged dimeric five coordinated Mg2 (1,2)2complex (Fig. 12).

Structure of Zn4(3)2(Me)2(OMe)2

Structure of Mg2(2)2
These compounds were screened for the polymerization of rac-LA in toluene at room temperature. The polymerization of rac-LA with the lithium complex Li4(3)2(THF) gave slightly atactic PLA (Pr = 0.49). The Mg(II), Zn(II) complexes in the presence of benzyl alcohol produced atactic PLA with narrow molecular weight distribution (Scheme 26).
Buchard et al. synthesized Nd(III), Sm(III) and Yb(III) complexes by using meso bispyrrolidine salan ligands with various lanthanide isopropoxide as the metal precursor [74]. The methyl substituted derivatives of bispyrrolidine salan favoured phenoxide bridged complexes of Sm (51%) and Yb (47%). On the other hand, the t-butyl substituted derivative prefers isopropoxide bridged complexes of Nd (86%) and Sm (7%). The lanthanide atom has a distorted octahedral geometry in the complex. The Sm(III) complex with t-butyl substituted bispyrrolidine salan was present as a mono-hydroxide species in the same solution which can be further hydrolyzed to form a bis-hydroxide dimer. Among the complexes with isopropoxide bridged structure, Nd(III) had the best control over PLA synthesis. The mono and di hydroxyl bridged complexes of Sm(III), and Yb(III) were not as active as its isopropoxide analogue due to the slow initiation behaviour of hydroxyl (–OH) group towards ROP (Scheme 27).

Lanthanum complexes of bispyrrolidine salan
Buchard and Jones reported a series of group 13 complexes from meso bispyrrolidine salan ligand [75]. The potassium salt of the ligand and along with group 13 metal halides were used for the preparation of Ga(III) and In(III) complexes. For the Al(III) complex, diethyl aluminium chloride was used as a precursor for Al(III). With the increase in the size of the metal ion, there was a switch over in the structure of the complexes from pseudo trigonal bipyramidal to square pyramidal. Both in the solution and solid phase, smaller Al(III) and Ga(III) complexes had pseudo trigonal bipyramidal geometry but the larger In(III) complex had square-based pyramidal geometry (Scheme 28).

Group 13 metal salan complexes
The Al(III) and Ga(III) complexes were inactive for the polymerization of rac-LA. The t-butyl group attached to the ligand and the small size of the metal ions did not allow the monomer to coordinate in an effective manner. However, the In(III) compound produced PLA with controlled molecular weight with very high heterotacticity (Pr = 0.82) in toluene at 80 °C since the larger size of In(III) allowed easy coordination to the LA monomer.
New Al(III) complexes with substituted and unsubstituted enantiomerically pure [(R,R) and (S,S)]bispyrrolidine salan and its rac-derivative [equimolar mixture of (R,R) and (S,S)] were reported by Kol (Scheme 29) [76]. The Al(III) complexes were synthesized with Me3Al and Et3Al respectively as the metal precursor. The t-butyl derivative of enantiomerically pure (R,R) bispyrrolidine salan formed a pentacoordinate complex on reaction with Et3Al which has a geometry in between trigonal bipyramidal and square pyramidal. The polymerization of rac-LA was carried with the [{ONNO}AlEt] complexes in the presence of benzyl alcohol in toluene at 50 °C and 70 °C respectively. The reaction of these complexes with benzyl alcohol first generated the corresponding alkoxide species that act as the active catalyst for the polymerization reactions. The polymerization of rac-LA at 70 °C with the aluminium complex of enantiopure (R,R) bispyrrolidine salan (Lig1H2) gave isotactic PLA (Pm = 0.72). The degree of isotacticity increased (Pm = 0.79) on lowering the polymerization temperature to 50 °C. However, the polymerization done with the aluminium complex associated to rac-derivative had less stereocontrol (Pm = 0.56 at 70 °C and Pm = 0.59 at 50 °C) compared to the complexes with enantiopure ligands. On the other hand, the chloro substituted enantiopure catalyst exhibited a moderate degree of heterotacticity (Pr = 0.64) and its rac-analogue gave a highly heterotactic PLA (Pr = 0.86) at 70 °C. On lowering the temperature to 50 °C the aluminium complex ligated to enantiopure ligand showed identical tacticity while for the rac- ligand, the aluminium complex gave highly heterotactic PLA (Pr ≥ 0.98). The formation of highly heterotactic PLA was explained by insertion, auto inhibition and polymeryl exchange mechanism. For the chiral salan, insertion of a specific LA enantiomer depends on a specific combination of salan chirality. If an inappropriate monomer is coordinated to the inappropriate metal enantiomer, it forms an inactive diastereomer. The ring opened monomer attached to the inactive salan transfers to the salan of opposite chirality via a presumable alkoxo bridged dinuclear species. It regenerates a diastereomer that has an active combination of salan and the last inserted LA monomer. This can now insert the LA enantiomer of opposite chirality to exhibit heterotactic PLA. The key step for this phenomenon is the auto inhibition process which constitutes the blocking of the catalytic site after LA insertion takes place, to exchange with the inappropriate polymeryl chain bound to the opposite salan aluminium enantiomer.

PLA tacticity from different salan aluminium complexes
Monopyrrolidine based Al(III) complexes were reported by Jones and co-workers. These compounds were screened towards the ROP of rac-LA [77]. In addition, they investigated the influence of the structural aspect of the ligand on the stereochemistry of the polymer. The different ligands were synthesized by reductive amination (a and e), through a modified Mannich condensation (b-d) and via an SN2 reaction (f) (Scheme 30). The aluminium complexes were prepared by the reaction of the ligands with 0.5 or 1 equivalent of Me3Al. Among the different possible aluminium complexes only the mono-ligated and bis-ligated complexes (shown in Scheme 30) could be isolated and characterized by single-crystal X-ray diffraction technique. The single crystal X-ray diffraction studies showed that in the mono-ligated complex, the Al(III) center is in a tetrahedral coordination environment and in the bis-ligated complex, the Al(III) center adopts pseudo trigonal bipyramidal geometry. A comparative study for bis-ligated monopyrrolidinealuminium complexes and bispyrrolidinealuminium complex earlier reported by the same group [67] revealed that in the monopyrrolidinebis-ligated complex [Al(X)2Me], the nitrogen centers are trans to each other while for the bipyrrolidine complexes, the nitrogen centers are cis to each other. Hence, there is a ring strain present in the bispyrrolidine complexes in the transition state during LA polymerization. As a result, the monopyrrolidine complexes react faster.

Synthesis of monopyrrolidine ligand and its Al(III) complexes
The polymerization reactions with bispyrrolidineAl(III) complex showed that the methyl substituted meso and enantiopure bispyrrolidineAl(III) complexes produced heterotactic and atactic PLA respectively while the t-butyl substituted derivative gave atactic PLA, indicating that the heterotactic nature of the polymer was influenced by the chirality of the ligand as well as the steric bulk of ortho substituent in the salan ligand. In addition, the polymerization of rac-LA with both mono- and bis-ligated, monopyrrolidine-based Al(III) complexes[Al(b)Me2 and Al(b)2Me] in the presence of benzyl alcohol produced isotactic biased block PLA with Pm = 0.80 The mono-ligated complexes react faster than their bis-ligated analogues towards ROP of rac-LA due to reduced steric hindrance around the metal center. Interestingly, all other Al(III) derivatives of mono- and bis-ligated monopyrrolidine ligand produced atactic PLA.
Sparteine: metal free catalyst for ROP
Sparteine is a tetracyclic plant alkaloid having the bis-quinolizidine moiety and is chiral [78] in nature. It is being used as a catalyst for the ROP of LA.
In 2017, Guo et al. reported a binary organocatalyst with guanidiumhexahydro-2H-pyrimido[1,2-α]pyrimidin-1-ium [(HppH2)+] as an ionic H-bond donor (iHBD) and tertiary amine sparteine (SP) as a H-bond acceptor (HBA) for the one-pot and two-step synthesis of polysarcosine-block-polylactide (PSar-b-PLA) diblock copolymer (Scheme 31) [79]. The first step of the synthesis involved the formation of polysarcosine (PSar) by the ROP of sarcosine N-carboxyanhydride initiated by benzylamine. In the second step, the H-bonding catalyst [(HppH2)+BF4−/ ( −)-sparteine] in conjugation with amine-terminated PSar, catalyzed the ROP of LA. The diblock copolymer (PSar-b-PLA) was characterized by NMR spectroscopy. The 1H NMR study was found to be consistent for the repeating structure of PSar. Again, for the PLA repeating block, 1H NMR signals were found at δ = 1.38 − 1.53 ppm and δ = 5.13 − 5.23 ppm respectively. The [(HppH2)+ BF4−] and [SP] combination is very efficient to produce amine initiated ROP of LA and the polysarcosine-b-polylactide diblock copolymer can be prepared with narrow dispersities around 1.16 − 1.21 and the experimental molecular weight was in good agreement with the theoretical molecular weight.

Synthesis of diblock copolymer PSar-b-PLA
Here is an example of the utilization of a hydrogen bond motif to get precise molecular mass polymer with narrow dispersity in dichloromethane at room temperature in the presence of benzyl alcohol as the initiator [80]. For the motif, different derivatives of squaramide [Sq1-3] as HBD (hydrogen-bond donor) and ( −)-sparteine as HBA (hydrogen-bond acceptor) were used (Scheme 32). [Sq1-3] alone cannot polymerize L-LA and the conversion rate with only ( −)-sparteine was very low. The percentage conversion of L-LA to PLLA [poly(L-lactide)] with three squaramide derivatives were Sq1: > 97%, Sq2: 53% and Sq3: 41% respectively. Thus, the best conversion rate was obtained with the electron-deficient squaramide and ( −)-sparteine due to strong hydrogen-bonding between the monomer and the organocatalyst. The ROP of L-LA with Sq1 as the hydrogen-bond donor, was more facile than with thiourea (conversion = 84%) under the same reaction conditions as Sq1 is better HBD than thiourea due to more acidic character of Sq1 as compared to thiourea. The ROP of L-LA with Sq1 and ( −)-sparteine was found to be living in nature. This was supported by the linear relationship between molecular weight and monomer conversion and a narrow PDI value of 1.06.

Different squaramide compounds and the mechanism for PLLA synthesis
For the mechanistic studies, a proton NMR titration was done by preparing a solution of benzyl alcohol and ( −)-sparteine in 1:1 ratio in CDCl3. There was a shift in the signal of the − OH proton from δ = 1.57 ppm to δ = 2.37 ppm as a result of hydrogen bonding with the tertiary amine, ( −)-sparteine. The signal of ethylene proton of BnOH shifted from δ = 4.63 ppm (d, J = 5.9 Hz) to δ = 4.66 ppm (s) indicating the formation of the hydrogen-bonded complex.
Subsequently, it was found for the first time that thiourea amine catalyst could be used for controlled ROP of L-LA [81]. An electron-withdrawing group attached to thiourea moiety acts as a better HBD due to the greater availability of labile hydrogen and the amine unit acts as the HBA. The HBD and HBA moieties of the thiourea amine catalyst can be present in the single-molecule (Scheme 33) or two different molecules (Scheme 34). A detailed study was carried out using a different structural variation of the thiourea amine catalyst towards the ROP of LA where the HBD and HBA units were not present in a single molecule. Different substituted thiourea moiety was prepared (Scheme 34) and screened for the polymerization of L-LA in the presence of t-amine NCyMe2 at room temperature with 4-pyrene-1-butanol as the initiator. The highest conversion to PLLA was achieved with thiourea having the most electron-withdrawing substituent (34a) (conversion = 98%) as electron-withdrawing groups enhance hydrogen-bonding ability of the thiourea moiety. The replacement of aliphatic cyclohexyl moiety by the aromatic ring (34 g and 34 h) gave nearly the same conversion as (34a), but the conversion decreased for (34 h) due to increase in the steric hindrance. Further studies were carried out using substituted thiourea (34a) with the variation of amines [pyridine, N,N-dimethylaniline, proton sponge, DABCO, TMEDA, triethylamine, DMAP, rac-TMCHD and ( −)-sparteine]. ( −)-sparteine was the most effective for activation of alcohol for both initiation and propagation with thiourea moiety towards the ROP of L-LA with 99% conversion achieved in 2 h. The reaction time reduced to about 25 folds with respect to the tertiary amine NCyMe2 since the availability of nitrogen electron density decreased due to the bulky cyclohexyl group present in NCyMe2 and which is absent in ( −)-sparteine.

HBD and HBA is in the same system

Different HBD and HBA systems
For the polymerization of rac-LA with (34a), ( −)-sparteine gave the best result with 99% conversion in 2 h and the polymer obtained was isotactic (Pm = 0.77).
Coulembier reported that ( +)-sparteine resulted in the controlled formation of cyclic polyester from L-LA under aprotic conditions [82]. The nitrogen atom in the sparteine is a very weak base. Hence, its nucleophilicity is sufficient for the ROP of L-LA. From the mechanism, it was found that the ROP of L-LA by nucleophilic nitrogen atom of ( +)-sparteine (Scheme 35) with subsequent backbiting reaction of LA gives a pure PLA macrocycle. There is a probability of ring-closure reaction at the zwitterionic nitrogen center, namely tail-biting (Scheme 36), which is the reason for the broad dispersity of the polymer. There were signals for the cyclic PLA in MALDI-TOF spectrum, separated by 72 mass units (mass of a half-LA unit). This indicates that both even and odd numbered macrocycles were formed.

( +)-Sparteine as a nucleophile

Mechanism for PLA macrocycle formation
In the presence of a protic solvent, only transesterification occurs since the protic solvent has better nucleophilicity than sparteine. Hence, controlling the backbiting reactions is possible under aprotic conditions.
In the same year, Bibal et al. exploited non-protonated ionic moieties as an alternative organocatalyst in place of classical hydrogen-bonding catalyst and the role of ion–dipole interaction for ROP was investigated [83]. They studied the influence of the quaternary ammonium moiety as HBDs to facilitate ROP. They synthesized different quaternary ammonium compounds (that served as multiple HBDs) (Scheme 37) from readily available nitrogen compounds with bis(trifluoromethane)sulfonamide[NTf2] or tetrakis[3,5-bis-(trifluoromethyl)phenyl]borate (BARF) as counter ions (Fig. 13). In addition, the sodium salt of [15-c-5] crown ether with NTf2 or BARF was used to study the role of cation-dipole interaction in the activation of the monomer (Scheme 38). For both the HBD and non-protonated ionic metal complex, HBA is required to trigger ROP of the monomer. ( −)-sparteine and DBU act as a HBA cocatalyst for the ROP of LA and lactone (ε-caprolactone and ẟ-valerolactone) respectively. For the polymerization of LA and lactones, the species paired with the BARF anion was more efficient than the pair with tight NTf2. For the polymerization of rac-LA, DBU-Me, DABCO-Me2, DMAP-Me and [15-c-5]Na gave almost complete conversion in 24 h. The conversion to PCL (polycaprolactone) was low even with long duration of time (120 h) with the best conversion (53%) obtained with [15-c-5]Na. Again, for the polymerization of ẟ-valerolactone, almost 100% conversion to the polymer was achieved in 12 h irrespective of the catalyst. The polymerization in all cases was well controlled yielding polymer with narrow PDI value ranging from 1.04 to 1.16.

Synthesis of quaternary ammonium catalysts

Structure of ionic catalysts and cocatalysts

Mechanism of ROP using crown ether and initiator
A photoswitchable catalyst was reported by Hecht et al. which interchanges as keto-enol form by tautomerism in different energies of light. The keto form which is predominant in ultraviolet light is inactive towards the ROP of L-LA and the enol form, predominant in the visible region can coordinate to the LA monomer via hydrogen-bonding and thus is active towards the ROP. By changing UV to visible light, easy switch over from keto to enol form was possible and the polymerization could be remotely controlled. The enol form (490 nm) is changed to the keto form (300 nm) in presence of N,N-dimethylcyclohexylamine. This catalytically off species was also characterized by X-Ray crystallography. ( −)-sparteine acts as a cocatalyst for the initiation and propagation of the ROP reaction. The photoswitchable catalyst and ( −)-sparteine together polymerized L-LA in a living manner (Scheme 39) [84].

Mechanismof PLA synthesis using photoswitchable catalyst
Azaphosphatrane was used as a HBD and its globular rigid structure causes steric hindrance which enhances the control of PLA synthesis by desired interaction with a HBA like ( −)-sparteine [85]. The different derivatives of azaphosphatrane were used along with bis(trifluoromethane)sulfonamide (NTf2−) and tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF−) as non-coordinating counterions (Scheme 40). These counter ions have strong hydrogen bonds with the azaphosphatrane unit, which makes the overall system a good HBD to LA. The X-ray crystallographic studies revealed that the phosphonium moiety in azaphosphatrane is located in the hydrophobic pocket on the top of the globular structure and P+ − H interact with the carbonyl group of the lactide monomer. With 10 mol % catalyst loading, the conversion to PLA reached 74 − 81%. Complete conversion was not achieved probably due to steric hindrance from azaphosphatrane that allows only moderate activation of the carbonyl group of rac-LA by the HBD. The hydrogen bonding between the monomer and azaphosphatrane moiety is weak and was supported by 1H and 31P NMR studies where the binding constant cannot be determined accurately due to small changes in the chemical shift values. The conversion percentage was independent of nature of the counter ion and substituent present in the azaphosphatrane unit.

ROP using Azaphosphatranes and ( −)-sparteine
Conclusion
Over the last decade, there has been significant research work dedicated to the synthesis of biodegradable polymers since conventional plastics are a threat to our environment and will eventually get exhausted in the long run due to depletion in petrochemical feedstocks. PLA, due to its biodegradable and biocompatible nature is an excellent alternative. The main aim of the synthetic pathway that governs the synthesis of PLA is to develop catalysts that can function under ambient condition, control tacticity, can be easily synthesised with no toxicity.
This review has considered the use of the macrocyclic moiety to develop both metal-based catalyst as well as organocatalyst towards the ROP of LA. The tuning of the parent macrocyclic moiety by suitable substitution and change in the metal center has a significant effect on the catalytic activity as well as tacticity of the polymer. This is well highlighted in this review. The major challenge is the focus of the extensive area of research to develop catalysts that work under ambient condition in very short time to give narrow dispersed PLA.
From the study we learn that, on increasing the rigidity of ligand framework from bispyrrolidine salan to porphyrin the controlled ability in the polymerization of LA (which is denoted by PDI value) by catalyst increases but the molecular weight of resulting polymer decreases. A synergy between the molecular weight and PDI of the PLA is crucial.
Change history
11 April 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10847-021-01060-y
Abbreviations
- ROP:
-
Ring Opening Polymerization
- LA:
-
Lactide
- PLA:
-
Polylactide
- PGA:
-
Polyglycolide
- PLGA:
-
Poly Lactic-co-Glycolic-acid
- PCL:
-
Polycaprolactone
- PDI:
-
Polydispersity index
- TPPH2 :
-
Tetraphenylporphyrin
- PO:
-
Propylene oxide
- PPO:
-
Polypropylene oxide
- PPN+Cl− :
-
Bis(triphenylphosphine) iminium chloride
- CHO:
-
Cyclohexene oxide
References
Chen, G.Q., Patel, M.K.: Plastics derived from biological sources: present and future: a technical and environmental review. Chem. Rev. 112, 2082–2099 (2012)
Dechy-Cabaret, O., Martin-Vaca, B., Bourissou, D.: Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 104, 6147–6176 (2004)
Rhim, J.W., Park, H.M., Ha, C.S.: Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 38, 1629–1652 (2013)
Hayashi, T.: Biodegradable polymers for biomedical uses. Prog. Polym. Sci. 19, 663–702 (1994)
Chiellini, E., Solaro, R.: Biodegradable polymeric materials. Adv. Mater. 8, 305–313 (1996)
Ikada, Y., Tsuji, H.: Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 21, 117–132 (2000)
Sinclair, R.G.: The case for polylactic acid as a commodity packaging plastic. J Mass Spec-Pure Appl Chem 33, 585–597 (1996)
Ahmed, J., Varshney, S.K.: Polylactides—chemistry, properties and green packaging technology: a review. Int. J. Food Prop. 14, 37–58 (2011)
Bogaert, J.C., Coszach, P.: Poly (lactic acids): a potential solution to plastic waste dilemma. Macromol. Symp. 153, 287–303 (2000)
Cheng, Y., Deng, S., Chen, P., Ruan, R.: Polylactic acid (PLA) synthesis and modifications: a review. Front Chem China 4, 259–264 (2009)
Hu, Y., Daoud, W.A., Cheuk, K.K.L., Lin, C.S.K.: Newly developed techniques on polycondensation, ring-opening polymerization and polymer modification: Focus on poly (lactic acid). Materials 9, 133 (2016)
Stanford, M.J., Dove, A.P.: Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 39, 486–494 (2010)
Sarazin, Y., Carpentier, J.F.: Discrete cationic complexes for ring-opening polymerization catalysis of cyclic esters and epoxides. Chem. Rev. 115, 3564–3614 (2015)
Dagorne, S., Fliedel, C.: Organoaluminum species in homogeneous polymerization catalysis. Top. Organomet. Chem. 41, 125–172 (2013)
Ghosh, S., Gowda, R.R., Jagan, R., Chakraborty, D.: Gallium and indium complexes containing the bis (imino) phenoxide ligand: synthesis, structural characterization and polymerization studies. Dalton Trans. 44, 10410–10422 (2015)
Mandal, M., Monkowius, U., Chakraborty, D.: Synthesis and structural characterization of titanium and zirconium complexes containing half-salen ligands as catalysts for polymerization reactions. New J. Chem. 40, 9824–9839 (2016)
O’Keefe, B.J., Hillmyer, M.A., Tolman, W.B.: Polymerization of lactide and related cyclic esters by discrete metal complexes. J. Chem. Soc. Dalton Trans. 15, 2215–2224 (2001)
Nederberg, F., Connor, E.F., Möller, M., Glauser, T., Hedrick, J.L.: New paradigms for organic catalysts: the first organocatalytic living polymerization. Angew. Chem. Int. Ed. 40, 2712–2715 (2001)
Zhang, L., Nederberg, F., Messman, J.M., Pratt, R.C., Hedrick, J.L., Wade, C.G.: Organocatalytic stereoselective ring-opening polymerization of lactide with dimeric phosphazene bases. J. Am. Chem. Soc. 129, 12610–12611 (2007)
Hormnirun, P., Marshall, E.L., Gibson, V.C., Pugh, R.I., White, A.J.P.: Study of ligand substituent effects on the rate and stereoselectivity of lactide polymerization using aluminumsalen-type initiators. Proc. Natl. Acad. Sci. U. S. A. 103, 15343–15348 (2006)
Qian, F., Liu, K., Ma, H.: Amidinate aluminium complexes: synthesis, characterization and ring-opening polymerization of rac-lactide. Dalton Trans. 39, 8071–8083 (2010)
Stasiw, D.E., Luke, A.M., Rosen, T., League, A.B., Mandal, M., Neisen, B.D., Cramer, C.J., Kol, M., Tolman, W.B.: Mechanism of the polymerization of rac-lactide by fast zinc alkoxide catalysts. Inorg. Chem. 56, 14366–14372 (2017)
Pedersen, C.J.: Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 89, 2495–2496 (1967)
Cram, D.J., Cram, J.M.: Host-guest chemistry. Science 183, 803–809 (1974)
Lehn, J.M.: Supramolecular Chemistry: Concepts and Perspectives. VCH, Weinheim (1995)
Battersby, A.R., Fookes, C.J., Matcham, G.W., McDonald, E.: Biosynthesis of the pigments of life: formation of the macrocycle. Nature 285, 17–21 (1980)
Mokhtari, B., Pourabdollah, K., Dallali, N.: A review of calixarene applications in nuclear industries. J. Radioanal. Nucl. Chem. 287, 921–934 (2011)
Nimse, S.B., Kim, T.: Biological applications of functionalized calixarenes. Chem. Soc. Rev. 42, 366–386 (2013)
Redshaw, C.: Coordination chemistry of the larger calixarenes. Coord. Chem. Rev. 244, 45–70 (2003)
O’Keefe, B.J., Hillmyer, M.A., Tolman, W.B.: Polymerization of lactide and related cyclic esters by discrete metal complexes. J. Chem. Soc. Dalton Trans. (2001). https://doi.org/10.1039/b104197p
Amgoune, A., Thomas, C.M., Carpentier, J.F.: Controlled ring-opening polymerization of lactide by group 3 metal complexes. Pure Appl. Chem. 79, 2013–2030 (2007)
Dos Santos Vieira, I., Herres-Pawlis, S.: Lactide Polymerisation with Complexes of Neutral N-Donors–New Strategies for Robust Catalysts. Eur. J. Inorg. Chem. 2012, 765–774 (2012)
Kremer, A.B., Mehrkhodavandi, P.: Dinuclear catalysts for the ring opening polymerization of lactide. Coord. Chem. Rev. 380, 35–57 (2019)
Sauer, A., Kapelski, A., Fliedel, C., Dagorne, S., Kol, M., Okuda, J.: Structurally well-defined group 4 metal complexes as initiators for the ring-opening polymerization of lactide monomers. Dalton Trans. 42, 9007–9023 (2013)
Wheaton, C.A., Hayes, P.G.: Designing cationic zinc and magnesium catalysts for coordination–insertion polymerization of lactide. Comments Inorg. Chem. 32, 127–162 (2011)
Chisholm, M.H.: Concerning the ring-opening polymerization of lactide and cyclic esters by coordination metal catalysts. Pure Appl. Chem. 82, 1647–1662 (2010)
Osten, K.M., Mehrkhodavandi, P.: Indium catalysts for ring opening polymerization: exploring the importance of catalyst aggregation. Acc. Chem. Res. 50, 2861–2869 (2017)
Sergeeva, E., Kopilov, J., Goldberg, I., Kol, M.: 2, 2′-Bipyrrolidine versus 1, 2-Diaminocyclohexane as Chiral Cores for Helically Wrapping Diamine−Diolate Ligands. Inorg. Chem. 48, 8075–8077 (2009)
Jiang, Z., Zhao, J., Zhang, G.: Ionic organocatalyst with a urea anion and Tetra-n-butyl ammonium cation for rapid, selective, and versatile ring-opening polymerization of lactide. ACS Macro Lett. 8, 759–765 (2019)
Engel, J., Cordellier, A., Huang, L., Kara, S.: Enzymatic Ring-Opening Polymerization of Lactones: Traditional Approaches and Alternative Strategies. ChemCatChem 11, 4983–4997 (2019)
Kamber, N.E., Jeong, W., Waymouth, R.M., Pratt, R.C., Lohmeijer, B.G.G., Hedrick, J.L.: Organocatalytic ring-opening polymerization. Chem. Rev. 107, 5813–5840 (2007)
Mezzasalma, L., Dove, A.P., Coulembier, O.: Organocatalytic ring-opening polymerization of L-lactide in bulk: A long standing challenge. Eur. Polym. J. 95, 628–634 (2017)
Pothupitiya, J.U., Dharmaratne, N.U., Jouaneh, T.M.M., Fastnacht, K.V., Coderre, D.N., Kiesewetter, M.K.: H-Bonding Organocatalysts for the Living, Solvent-Free Ring-Opening Polymerization of Lactones: Toward an All-Lactones. All-Conditions Approach. Macromolecules 50, 8948–8954 (2017)
Lin, B., Waymouth, R.M.: Organic ring-opening polymerization catalysts: reactivity control by balancing acidity. Macromolecules 51, 2932–2938 (2018)
Ottou, W.N., Sardon, H., Mecerreyes, D., Vignolle, J., Taton, D.: Update and challenges in organo-mediated polymerization reactions. Prog. Polym. Sci. 56, 64–115 (2016)
Zhang, D., Zi, G.: N-heterocyclic carbene (NHC) complexes of group 4 transition metals. Chem. Soc. Rev. 44, 1898–1921 (2015)
Wink, M., Meißner, C., Witte, L.: Patterns of quinolizidine alkaloids in 56 species of the genus Lupinus. Phytochemistry 38, 139–153 (1995)
Tempelaar, S., Mespouille, L., Dubois, P., Dove, A.P.: Organocatalytic synthesis and postpolymerization functionalization of allyl-functional poly (carbonate) s. Macromolecules 44, 2084–2091 (2011)
Mostovaya, O.A., Gorbachuk, V.V., Padnya, P.L., Vivilova, A.A., Evtugyn, G.A., Stoikov, I.I.: Modification of oligo- and polylactides with macrocyclic fragments: synthesis and properties, p. 7. Front, Chem (2019)
Gorbachuk, V.V., Padnya, P.L., Mostovaya, O.A., Gerasimov, A.V., Stoikov, I.I.: Towards novel functional polymers: Ring-opening polymerization of L-lactide with p-tert-butylthiacalix[4]arene derivatives. React. Funct. Polym. 150, 104546 (2020)
Dubois, P., Jacobs, C., Jérôme, R., Teyssie, P.: Macromolecular engineering of polylactones and polylactides. 4. Mechanism and kinetics of lactide homopolymerization by aluminumisopropoxide. Macromolecules 24, 2266–2270 (1991)
Imran, M., Ramzan, M., Qureshi, A.K., Khan, M.A., Tariq, M.: Emerging applications of porphyrins and metalloporphyrins in biomedicine and diagnostic magnetic resonance imaging. Biosensors 8, 95 (2018)
Trofimoff, L., Aida, T., Inoue, S.: Formation of poly (lactide) with controlled molecular weight. Polymerization of lactide by aluminum porphyrin. Chem. Lett. 16, 991–994 (1987)
Li, D., Gao, B., Duan, Q.: Syntheses of biodegradable and biorenewable polylactides initiated by aluminum complexes bearing porphyrin derivatives by the ring-opening polymerization of lactides. J Biomat Sci-Polym E 30, 846–860 (2019)
Balasanthiran, V., Chatterjee, C., Chisholm, M.H., Harrold, N.D., RajanBabu, T.V., Warren, G.A.: Coupling of propylene oxide and lactide at a porphyrin chromium (III) center. J. Am. Chem. Soc. 137, 1786–1789 (2015)
Summerville, D.A., Jones, R.D., Hoffman, B.M., Basolo, F.: Chromium (III) porphyrins. Chemical and spectroscopic properties of chloro-meso-tetraphenylporphinatochromium (III) in nonaqueous solutions. J. Am. Chem. Soc. 99, 8195–8202 (1977)
Chatterjee, C., Chisholm, M.H.: Ring-Opening Polymerization Reactions of Propylene Oxide Catalyzed by Porphyrin Metal (3+) Complexes of Aluminum. Chromium and Cobalt. Chem. Rec. 13, 549–560 (2013)
Anker, M., Balasanthiran, C., Balasanthiran, V., Chisholm, M.H., Jayaraj, S., Mathieu, K., Piromjitpong, P., Praban, P., Raya, B., Simonsick, W.J.: A new route for the preparation of enriched iso-polylactide from rac-lactide via a Lewis acid catalyzed ring-opening of an epoxide. Dalton Trans. 46, 5938–5945 (2017)
Praban, S., Piromjitpong, P., Balasanthiran, V., Jayaraj, S., Chisholm, M.H., Tantirungrotechai, J., Phomphrai, K.: Highly efficient metal (III) porphyrin and salen complexes for the polymerization of rac-lactide under ambient conditions. Dalton Trans. 48, 3223–3230 (2019)
Li, D., Gao, B., Duan, Q.: Preparation of star-shaped functionalized polylactides by metal porphyrin complexes as both catalysts and cocatalysts. J. Porphyrins Phthalocyanines 23, 1020–1027 (2019)
Li, Y., Zhao, K.Q., Redshaw, C., Ortega, B.A.M., Nuñez, A.Y., Hanna, T.A.: Coordination chemistry and applications of phenolic calixarene–metal complexes. PATAI’S Chemistry of Functional Groups (2009). https://doi.org/10.1002/9780470682531.pat0616
Bukhaltsev, E., Frish, L., Cohen, Y., Vigalok, A.: Single-site catalysis by bimetallic zinc calixarene inclusion complexes. Org. Lett. 7, 5123–5126 (2005)
Frediani, M., Sémeril, D., Mariotti, A., Rosi, L., Frediani, P., Rosi, L., Matt, D., Toupet, L.: Ring Opening Polymerization of Lactide under Solvent-Free Conditions Catalyzed by a Chloro titanium Calix [4] arene Complex. Macromol. Rapid Commun. 29, 1554–1560 (2008)
Frediani, M., Sémeril, D., Matt, D., Rosi, L., Frediani, P., Rizzolo, F., Papini, A.M.: Ring-opening polymerisation of rac-lactide using a calix 4 arene-based titanium (IV) complex. Int J. Polym. Sci (2010). https://doi.org/10.1155/2010/490724
Walton, M.J., Lancaster, S.J., Redshaw, C.: Highly Selective and Immortal Magnesium Calixarene Complexes for the Ring-Opening Polymerization of rac-Lactide. ChemCatChem 6, 1892–1898 (2014)
Mayilmurugan, R., Traar, P., Schachner, J.A., Volpe, M., Mösch-Zanetti, N.C.: Dioxidomolybdenum (VI) Complexes Containing Ligands with the Bipyrrolidine Backbone as Efficient Catalysts for Olefin Epoxidation. Eur. J. Inorg. Chem. 2013, 3664–3670 (2013)
Hador, R., Botta, A., Venditto, V., Lipstman, S., Goldberg, I., Kol, M.: The Dual-Stereocontrol Mechanism: Heteroselective Polymerization of rac-Lactide and Syndioselective Polymerization of meso-Lactide by Chiral AluminumSalan Catalysts. Angew. Chem. Int. Ed. 58, 14679–14685 (2019)
Jones, M.D., Hancock, S.L., McKeown, P., Schäfer, P.M., Buchard, A., Thomas, L.H., Lowe, J.P.: Zirconium complexes of bipyrrolidine derived salan ligands for the isoselective polymerisation of rac-lactide. Chem. Commun. 50, 15967–15970 (2014)
Kaminsky, W., Arndt, M.: 17. Mechanism of the First Steps of the Isotactic Polymerization with Metallocene Catalysts. In: Kaminsky, W. (ed.) Studies in Surface Science and Catalysis, vol. 89, pp. 179–192. Elsevier, Amsterdam (1994)
Jones, M.D., Brady, L., McKeown, P., Buchard, A., Schäfer, P.M., Thomas, L.H., Lowe, J.P.: Metal influence on the iso-and hetero-selectivity of complexes of bipyrrolidine derived salan ligands for the polymerisation of rac-lactide. Chem. Sci. 6, 5034–5039 (2015)
Addison, A.W., Rao, T.N., Reedijk, J., van Rijn, J., Verschoor, G.C.: Synthesis, structure, and spectroscopic properties of copper (II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua [1, 7-bis (N-methylbenzimidazol-2′-yl)-2, 6-dithiaheptane] copper (II) perchlorate. J. Chem. Soc. Dalton Trans. (1984). https://doi.org/10.1039/DT9840001349
Yang, L., Powell, D.R., Houser, R.P.: Structural variation in copper (I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ 4. Dalton Trans (2007). https://doi.org/10.1039/B617136B
Quilter, H.C., Drewitt, R.H., Mahon, M.F., Kociok-Köhn, G., Jones, M.D.: Synthesis of Li (I), Zn (II) and Mg (II) complexes of amine bis (phenolates) and their exploitation for the ring opening polymerisation of rac-lactide. J. Organomet. Chem. 848, 325–331 (2017)
Beament, J., Kociok-Köhn, G., Jones, M.D., Buchard, A.: Bipyrrolidinesalan alkoxide complexes of lanthanides: synthesis, characterisation, activity in the polymerisation of lactide and mechanistic investigation by DOSY NMR. Dalton Trans. 47, 9164–9172 (2018)
Beament, J., Mahon, M.F., Buchard, A., Jones, M.D.: Salan group 13 complexes–structural study and lactide polymerisation. New J. Chem. 41, 2198–2203 (2017)
Press, K., Goldberg, I., Kol, M.: Mechanistic insight into the stereochemical control of lactide polymerization by salan–aluminum catalysts. Angew. Chem. Int. Ed. 54, 14858–14861 (2015)
Beament, J., Mahon, M.F., Buchard, A., Jones, M.D.: Aluminum complexes of monopyrrolidine ligands for the controlled ring-opening polymerization of lactide. Organometallics 37, 1719–1724 (2018)
Villalpando-Vargas, F., Medina-Ceja, L.: Sparteine as an anticonvulsant drug: Evidence and possible mechanism of action. Seizure 39, 49–55 (2016)
Chen, S., Liu, Y., Li, Z., Wang, X., Dong, H., Sun, H., Yang, K., Gebru, H., Guo, K.: H-bonding binary organocatalysis promoted amine-initiated ring-opening polymerizations of lactide from polysarcosine to diblock copolymers. Eur. Polym. J. 97, 389–396 (2017)
Liu, J., Chen, C., Li, Z., Wu, W., Zhi, X., Zhang, Q., Wu, H., Wang, X., Cui, S., Guo, K.: A squaramide and tertiary amine: an excellent hydrogen-bonding pair organocatalyst for living polymerization. Polym. Chem. 6, 3754–3757 (2015)
Pratt, R.C., Lohmeijer, B.G.G., Long, D.A., Lundberg, P.N.P., Dove, A.P., Li, H., Wade, C.G., Waymouth, R.M., Hedrick, J.L.: Exploration, Optimization, and Application of Supramolecular Thiourea−Amine Catalysts for the Synthesis of Lactide (Co) polymers. Macromolecules 39, 7863–7871 (2006)
Coulembier, O., De Winter, J., Josse, T., Mespouille, L., Gerbaux, P., Dubois, P.: One-step synthesis of polylactide macrocycles from sparteine-initiated ROP. Polym. Chem. 5, 2103–2108 (2014)
Thomas, C., Milet, A., Peruch, F., Bibal, B.: Activation of carbonyl bonds by quaternary ammoniums and a (Na+: crown-ether) complex: investigation of the ring-opening polymerization of cyclic esters. Polym. Chem. 4, 3491–3498 (2013)
Eisenreich, F., Kathan, M., Dallmann, A., Ihrig, S.P., Schwaar, T., Schmidt, B.M., Hecht, S.: A photoswitchable catalyst system for remote-controlled (co) polymerization in situ. Nat. Catal. 1, 516–522 (2018)
Zhang, D., Jardel, D., Peruch, F., Calin, N., Dufaud, V., Dutasta, J.P., Martinez, A., Bibal, B.: Azaphosphatranes as Hydrogen-Bonding Organocatalysts for the Activation of Carbonyl Groups: Investigation of Lactide Ring-Opening Polymerization. Eur. J. Org. Chem. 2016, 1619–1624 (2016)
Funding
Funding was provided by UGC-ISF Grant No. 6-2/2018(IC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective Open Access cancellation.
Rights and permissions
About this article

Cite this article
Roy, S.S., Sarkar, S. & Chakraborty, D. Macrocycles in dual role: ancillary ligands in metal complexes and organocatalysts for the ring-opening polymerization of lactide. J Incl Phenom Macrocycl Chem 100, 1–36 (2021). https://doi.org/10.1007/s10847-021-01045-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-021-01045-x