
Abstract
Three novel C4 symmetric O-substituted derivatives of tetramethoxyresorcinarene were synthesised and characterised and the selective distal lithiation of these derivatives was investigated. Lithiation of the tetraethoxy derivative, followed by quenching with dimethyldisulfide selectively afforded the distal product in 36% yield after removal of unreacted starting resorcinarene. Lithiation of the MOM derivative produced the five possible lithiated products, with the mono being the major product; the distally substituted derivative being a minor product. The benzyloxyresorcinarene appeared to be unstable under the lithation conditions. These results suggest that the selectivity of the distal lithation reaction is heavily dependent on the O-substituents of the resorcinarene.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Resorcinarenes are a sub-class of calixarenes and have been relatively less-studied compared to the mainstream calixarenes. Tetramethoxyresorcinarene is a type of resorcinarene which only has four hydroxyl groups instead of eight. It can be conveniently prepared in a single step in high yields [1]. The methoxy groups are arranged about the resorcinarene in an alternating manner, giving the resorcinarene C4 rotational symmetry, as well as helical chirality [2]. This helical chirality at the wider rim renders the cavity of the resorcinarene to be chiral. A chiral cavity is of significance because it has the potential for enantioselective containment of chiral guests. By adjusting the shape and functional groups at the wider rim of the cavity, this potential could be realised, which could lead to enantiorecognition applications. An interesting modification would be the functionalisation of the distal aromatic rings. This would enable interesting architectures to be constructed over the chiral cavity that would change its structure, and perhaps lead to enhanced containment of guests.
In general, the distal functionalisation of calixarenes is standard practice [3,4,5], but despite the similarities, for resorcinarenes it is far less common. This is because the hydroxyl groups in calixarenes, being on the narrower rim, are in proximity to each other, and hence the alkylation of the first hydroxyl causes the distal hydroxyl to be the most favourable for subsequent deprotonation because the resultant phenoxide is stabilised by two hydrogen bonds formed from the two proximal hydroxyl groups. Therefore, the second alkylation occurs on the distal hydroxyl [5]. Unfortunately this principle does not apply to resorcinarenes because of the innately different positions of the hydroxyl groups.
In the literature, there exists a few procedures for distally-functionalising resorcinarenes. In the method by Shivanyuk et al. [6, 7], a distally-functionalised resorcinarene was directly obtained in 53% yield through regioselective tetratosylation of octahydroxyresorcinarene. Under these conditions, octahydroxyresorcinarene was treated with triethylamine followed by four equivalents of tosyl chloride in dry acetonitrile. This resulted in the tosylation of the four hydroxyl groups of the distal aromatic rings, while the other pair of aromatic rings were left intact. Selectivity was likely to due to the observed precipitate which formed when the triethylamine was added to the resorcinarene. Addition of the sulfonylating agent to the precipitate needed to be performed quickly and followed by rapid stirring of the reaction mixture to avoid the formation of complicated mixtures. However, this method excludes the tetramethoxyresorcinarene because four of the hydroxyl groups have already been alkylated.
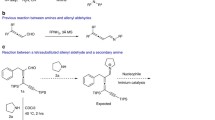
Aside from this work, in the literature there is one other main strategy for distally-functionalising a resorcinarene—that is by selective lithiation as reported by Arnott et al. [8] This direct lithiation method is similar to the selective bromine–lithium exchange by Larsen and Jørgensen on calixarenes [4] as well as by Sherburn on cavitands [9]. But in contrast, the lithiation by Arnott et al. is not a bromine–lithium exchange, and thus does not require a brominated resorcinarene. In this direct lithiation method, octamethoxyresorcinarene was lithiated using n-butyllithium, then quenched with dimethyldisulfide to give a thioether, which enabled elucidation of the result by 1H NMR spectroscopy of the crude mixture. The lithation under various conditions was investigated with variation of the reaction solvent, number of equivalents of butyllithium, lithiation time and temperature. Using five equivalents of n-butyllithium in THF at a temperature of 40 °C for 2 h, the best yield of 86% for the distal product was achieved, albeit the presence of unreacted starting material and mono product. The lithation under these optimal conditions was also demonstrated to work with other electrophiles, such as carbon dioxide, to give useful functional groups, while still retaining a good yield for the distal product. It was hypothesised that the distal selectivity was the result of the randomly-lithiated species equilibrating to the distally-lithiated intermediate which was presumably the most thermodynamically stable. Here we report our investigations into the applicability of this distal lithiation on a C4-symmetrical tetramethoxyresorcinarene.
Results and discussion
The first step to prepare tetramethoxyresorcinarene for the lithiation is to protect all four hydroxyl groups with a protecting group that would be stable under the lithation conditions (Scheme 1).

Investigation into the distal-lithiation of various O-protected resorcinarene derivatives
The protecting group first chosen was the benzyl ether. Benzyl ethers are known to be fairly robust, pH stable, and are usually only cleavable by reduction to return the alcohol. Furthermore, benzyl bromide is relatively safe to use and was readily available. Reaction of the starting resorcinarene (1) with benzyl bromide and sodium hydride in THF furnished the desired tetrabenzyloxyresorcinarene (2) in a good yield and purity after recrystallisation (Scheme 2). Evidence of the benzyl ether was provided by NMR spectroscopy, which showed the benzylic protons as an AB pattern at 4.70 and 4.94 ppm on the 1H spectrum, while the benzylic carbon appeared on the 13C spectrum at 71.1 ppm.

Protection of resorcinarene phenols with benzyl ethers and attempted distal lithiation
With tetrabenzyloxyresorcinarene (2) in hand, the lithiation was then performed according to the same conditions as described by Arnott et al., and quenching with dimethyldisulfide. Unfortunately, TLC did not show any well-defined spots and the 1H NMR spectrum of the quenched reaction mixture indicated the resorcinarene peaks as broad humps, with multiple methoxy and aromatic signals. The benzyl ether was still apparent by a broad hump from 6.7 to 7.7 ppm. This complex mixture perhaps suggests that the benzyl ether was not as robust as thought. A protecting group that would be more stable under the strongly basic lithiation conditions may perhaps provide success.
The methoxy methyl (MOM) protecting group, when functionalised to a phenol, is known to direct lithiation to the ortho position of the aromatic ring [10, 11]. MOM protecting groups can be easily removed by acid-catalysed hydrolysis. Therefore, the MOM protecting group is an ideal candidate for this application. However, the main issue is the safety of methoxymethyl halides [12], which is required for synthesising the MOM ether of the phenol. Nevertheless, the reaction of (1) with methoxymethyl bromide under standard sodium hydride/THF conditions afforded the target product (3) in excellent purity in quantitative yield without any formal purification (Scheme 3). NMR spectroscopy confirmed the MOM ether resorcinarene by an additional methoxy peak appearing on both 1H and 13C spectra. The acetal was evidenced by the AB pattern at 4.84 and 4.73 ppm on the 1H spectrum, as well as the CH2 peak at 95.8 ppm on the DEPT-135 spectrum.

Protection of resorcinarene phenols with methoxymethyl ethers and attempted distal lithiation
The lithiation was performed on resorcinarene (3) as per the literature, but yielded a mixture of six compounds as indicated by TLC (Rf(EtOAc/Petrol 4:6) = 0.71, 0.63, 0.48, 0.38, 0.31, 0.19, Fig. S1). Doubling the amount of n-butyllithium from five to ten equivalents also produced the same result, but with the least retained spot on TLC becoming darker. The crude products from the reactions with five and ten equivalents were combined, and the six compounds were separated sufficiently by preparative TLC to enable identification of each compound by 1H NMR spectroscopy. The structures of the six compounds were tentatively assigned by analysis of their 1H NMR spectra, based principally on symmetry and integration data. The 1H NMR data suggested that the six compounds obtained were the five possible products plus the starting material (Scheme 4). The 1H NMR data, together with the isolated yields and assignments are summarised in Table 1.

Lithiation of MOM resorcinarene produces the five possible SCH3 resorcinarene products
The signals in the 1H NMR spectra, which provided clear indicators for assigning the products were the SCH3 and the aromatic signals. The distal product (5), having C2 rotational symmetry, would have half the peaks compared to its asymmetrical proximal isomer (6). The appearance of many peaks indicated an absence of rotational symmetry in the product. These asymmetrical products—tri (7), proximal (6), and mono (4)—could be distinguished from each other by the number of ArH and SCH3 signals. For example, tri (7), having three SCH3 replacing three ArH, would have three less ArH from the original eight, which would result in five ArH peaks. Therefore, based on the number and integration of these signals, together with symmetry considerations, the particular SCH3 resorcinarene product could be assigned for each spectrum. For all isolated products, the retention of the MOM protecting group through the lithiation was confirmed by the pairs of AB doublets around 4.2–5.2 ppm and the corresponding extra methoxy singlet in all 1H NMR spectra.
It is clear from the results that distal di-SMe resorcinarene is not the main product. In efforts to optimise the yield for the target distal product, the lithiation was attempted under milder conditions, at room temperature overnight, but only returned unreacted starting resorcinarene. The lithiation was also attempted with sec-butyllithium, a stronger base, but yielded similar results as with n-butyllithium. From the results, it is clear that the lithiation of resorcinarene methoxymethyl ether (3) is not a viable method for obtaining a distally-functionalised resorcinarene due to the lack of selectivity and difficulty of separation.
Finally, for the purpose of simply testing the applicability of the lithation procedure on this particular tetramethoxyresorcinarene, the ethyl ether was chosen. The ethyl ether would not be readily cleavable to recover the phenol and has no functional value. Furthermore, as the methyl and ethyl groups are of similar size, the chirality influence would be minimal. However, it was for this similarity that the ethyl ether was chosen; to liken the resorcinarene to the literature octamethoxy resorcinarene, while retaining its characteristic C4 symmetry.
The tetraethylation of resorcinarene (1) under standard sodium hydride/THF conditions did not proceed to completion despite using up to 30 equivalents of iodoethane and subjecting the mixture to reaction again. However, changing the reaction solvent to DMF conveniently furnished the target product (10) in good yield and purity (Scheme 5). The ethoxy group was confirmed by 1H NMR spectroscopy as a multiplet overlapped with the methoxy singlet, and an additional triplet in the hydrocarbon region. The methyleneoxy carbon was accounted for on the DEPT-135 spectrum by the peak at 64.9 ppm with negative phase.

Ethylation of resorcinarene phenols and subsequent distal lithiation
The ethoxy derivative (10) was then subjected to the same lithiation procedure to give a single product along with unreacted starting resorcinarene, as indicated by TLC. The product was successfully separated by column chromatography to afford the target product (9) in yield of 36%. 1H NMR spectroscopy of the pure product conclusively showed a C2-symmetrical resorcinarene product with a single SCH3 peak at 2.42 ppm and three peaks in the aromatic region with the same integration. The correct number of peaks for target product (9) was present on the 13C NMR spectrum, with the SCH3 peak being identified as the peak at 18.3 ppm according to HSQC spectroscopy. Therefore, this experiment with the ethoxy functionalised resorcinarene proved that the lithiation for octamethoxyresorcinarene was applicable to this chiral resorcinarene, although the phenols needed to be alkylated with a group with close likeness to a methoxy group.
Conclusion
The selective distal lithiation of three novel C4 symmetric O-substituted derivatives of tetramethoxyresorcinarene was investigated. These included the benzyloxy, the MOM and the ethoxy derivatives of tetramethoxyresorcinarene. Lithiation, followed by quenching with dimethyldisulfide gave very different results for the three derivatives. The benzyloxy derivative gave a complex mixture that could not be resolved. The lithiation on the MOM ether resorcinarene (3) gave a mixture of the five possible products, as well as unreacted starting resorcinarene. Separation of the five products was not complete, but the isolated compounds were sufficiently pure for tentative characterisation. The most abundant product was the mono-SMe (4) which was recovered in approximately 28% yield, while the distal-SMe (5) was recovered in a minor yield of about 4%. The lithiation with ethoxyresorcinarene (10) selectively produced the distal-SMe product (9) in 36%, after separation of unreacted starting resorcinarene. This served as a proof of concept that the selective distal lithiation was applicable to C4 symmetric tetramethoxyresorcinarene, albeit with an O-substituent with close similarity to a methoxy. These results suggest that the selective distal lithiation is not very robust, with the distal selectivity being dramatically affected by the O-substituents on the resorcinarene.
Experimental
14,36,56,76-Tetrahydroxy-16,34,54,74-tetramethoxy-2,4,6,8-tetrapropylresorcin[4]arene (1)

Resorcinarene (1) was synthesised based on the procedure by McIldowie et al. [1]. To a mixture of 3-methoxyphenol (30.1 g, 0.243 mol), butanal (22 mL, 17.6 g, 0.245 mol) and DCM (550 mL) was added boron trifluoride etherate (90 mL, 104 g, 0.729 mol) while in an ice bath. The solution turned from orange–yellow to dark red. The ice bath was removed and the reaction mixture was allowed to warm to RT over about 20 min, while stirring under nitrogen. After 2 h at RT, the reaction mixture was quenched by adding water (400 mL), producing some white fumes and slight warming. The organic layer was separated, and the solvent was removed under reduced pressure to give a foamy brown solid as the crude product. The crude product was triturated with methanol (~ 200 mL) to give the desired product (1) (36.9 g, 85%) as a white powder: mp 256–259 °C (lit. [13] 257–258 °C); IR 3400 cm−1 (OH phenol); 1H NMR (CDCl3) δ 0.97 (t, J = 7.4 Hz, 12 H, CH2CH3), 1.30 (apparent sxt, 8 H, CH2CH3), 2.18 (apparent q, 8 H, CH2CH), 3.83 (s, 12 H, OCH3), 4.30 (t, J = 7.9 Hz, 4 H, CHCH2), 7.23 and 6.34 (2s, 2 × 4 H, ArH), 7.50 (s, 4 H, OH).
General reaction procedure for O-alkylation of resorcinarene (1)
Resorcinarene (1) (1 equiv) was added to a mixture of washed sodium hydride (10–20 equiv, 60% in oil) and a couple of crystals of imidazole in anhydrous THF, and the resultant mixture was stirred under nitrogen at RT for 30 min. To this white slurry was added the relavant alkyl halide (10–20 equiv), and the cloudy white reaction mixture was stirred overnight at RT, under nitrogen. The work up and purification is specific for each resorcinarene derivative.
14,36,56,76-Tetrabenzoxy-16,34,54,74-tetramethoxy-2,4,6,8-tetrapropylresorcin[4]arene (2)

The general reaction procedure was applied with resorcinarene (1) (0.50 g, 0.703 mmol), sodium hydride (1.07 g, 60% in oil, 0.64 g, 26.8 mmol), anhydrous THF (25 mL), and benzyl bromide (1.69 mL, 1.21 g, 14.0 mmol). After stirring overnight, the reaction mixture was cooled in an ice bath and carefully quenched with water, producing bubbles. More water was added, and the solvents were removed under reduced pressure (rotavap up to 65 °C). The yellow residue was dissolved in DCM, and the organic layer was washed with water, separated and evaporated to give the crude product as a yellow crystalline solid (0.88 g). The crude product was dissolved in minimum DCM; then MeOH was added so that the product remained dissolved. The yellow solution was gently boiled to remove DCM. At a certain point, white crystals formed in the solution, and the mixture was immediately taken off the heat and allowed to cool to RT. More MeOH was added, and (2) was collected by vacuum filtration as white crystals (0.64 g, 84%): mp 188–189 °C (DCM/MeOH); 1H NMR (CDCl3) δ 0.91 (t, J = 7.3 Hz, 12 H, CH2CH3), 1.31 (apparent sxt, 8 H, CH2CH3), 1.87 (m, 8 H, CH2CH), 3.40 (s, 12 H, OCH 3 ), 4.59 (t, J = 7.4 Hz, 4 H, CHCH2), 4.70, 4.95 (2 × AB, J = 11.3 Hz, 8 H, OCH2), 6.37, 6.71 (s, 2 × 4 H, ArH), 7.39–7.18 (m, 20 H, Ph); 13C NMR (CDCl3) δ 14.5 (CH2CH3), 21.6 (CH2), 35.9 (CHCH2), 37.1 (CH2CH), 55.6 (OCH3), 71.1 (OCH2), 97.8, 126.4 (CH, Ar), 126.5 (C, Ar), 127.5, 127.6, 128.3 (CH, Ar), 137.9, 155.2, 155.8 (C, Ar) (note some signals are coincident). Found: C, 80.58; H, 7.69; C72H80O8; requires C, 80.56; H, 7.51%.
14,36,56,76-Tetramethoxymethyl-16,34,54,74-tetramethoxy-2,4,6,8-tetrapropylresorcin[4]arene (3)

The general reaction procedure was applied with resorcinarene (1) (1.00 g, 1.40 mmol), sodium hydride (0.68 g, 60% in oil, 0.409 g, 17.0 mmol), anhydrous THF (8 mL), and bromomethyl methyl ether (0.345 mL, 1.76 g, 14.1 mmol). After stirring overnight, the reaction mixture was then quenched with methanol (2 mL) till bubbling ceased, and was then stirred for 45 min at RT. The solvent was removed under reduced pressure, and the residue was dissolved in DCM (30 mL) and dilute NaOH solution (20 mL, 1 M) to give a cloudy light-yellow organic layer and a clearer dark yellow aqueous layer. The layers were separated, and the aqueous layer was extracted with DCM (10 mL) to give a clear colourless extract. The combined cloudy organic extracts were dried (MgSO4), and solvent removed under reduced pressure to give pure (3) as a beige-coloured solid (1.36 g, ~ 100%): mp 149–150 °C (CHCl3/MeOH); 1H NMR (CDCl3) δ 0.92 (t, J = 7.3 Hz, 12 H, CH2CH3), 1.35 (apparent sxt, 8 H, CH2CH3), 1.82 (apparent q, 8 H, CH2CH), 3.34 and 3.63 (2s, 2 × 12 H, OCH 3 ), 4.51 (t, J = 7.5 Hz, 4 H, CHCH2), 4.73, 4.85 (2 × AB, 8 H, J = 6.4 Hz, OCH2O), 6.48, 6.66 (s, 2 × 4 H, ArH); 13C NMR (CDCl3) δ 14.4 (CH2CH3), 21.3 (CH2CH3), 35.3 (CH2CH), 37.4 (CHCH2), 55.7, 55.8 (OCH3), 95.8 (OCH2O), 100.1, 126.1 (CH, Ar), 127.0, 127.5, 153.6, 155.6 (C, Ar). Found: C, 70.16; H, 8.19; C52H72O12; requires C, 70.24; H, 8.16%.
14,36,56,76-Tetraethoxy-16,34,54,74-tetramethoxy-2,4,6,8-tetrapropylresorcin[4]arene (10)

The general reaction procedure was applied with resorcinarene (1) (0.101 g, 0.142 mmol), sodium hydride (0.103 g, 60% in oil, 0.062 g, 2.58 mmol), anhydrous DMF (25 mL), and iodoethane (0.168 mL, 0.328 g, 2.10 mmol). After stirring overnight, the cloudy reaction mixture was quenched with water, producing bubbles. Then water (50 mL) was added, the white precipitate was filtered, and washed with more water. The filtered white precipitate was washed off the funnel with DCM, and the solvent was removed under reduced pressure. The resultant residue was placed in an oven (140 °C) for 5 min to remove water, and furnish pure (10) as a white solid (0.106 g, 92%): mp 206–208 °C (EtOAc); 1H NMR (CDCl3) δ 0.93 (t, J = 7.3 Hz, 12 H, CH2CH2CH3), 1.18 (t, J = 7.0 Hz, 12 H, OCH2CH3), 1.37 (m, 8 H, CH2CH3), 1.82 (apparent q, 8 H, CH2CH), 3.59 (s, 12 H, ArOCH 3 ), 3.60–3.69 and 3.85–3.98 (2 m, 2 × 4 H, OCH2CH3), 4.51 (t, J = 7.5 Hz, 4 H, CHCH2), 6.29, 6.63 (s, 2 × 4 H, ArH); 13C NMR (CDCl3) δ 14.5, 15.0 (CH2CH3), 21.3 (CH2CH3), 35.2 (CHCH2), 37.2 (CH2CH), 55.9 (OCH3), 64.9 (OCH2), 98.1, 126.1 (CH, Ar), 126.6, 126.8, 155.3, 155.7 (C, Ar). Found: C, 75.66; H, 9.11; C72H80O8; requires C, 75.69; H, 8.80%.
Attempted selective distal lithiation of 14,36,56,76-tetrabenzyloxy-16,34,54,74-tetramethoxy-2,4,6,8-tetrapropylresorcin[4]arene
The synthesis was performed according to the procedure by Arnott et al. [8] To a clear colourless solution of resorcinarene benzyl ether (2) (0.050 g, 0.0503 mmol) in anhydrous THF (5 mL) at RT was added n-butyllithium (0.16 mL, 1.6 M, 0.256 mmol). The clear colourless solution immediately turned bright yellow, then dark yellow, then orange, then dark orange. The reaction mixture was stirred at 40 °C under nitrogen for 2 h. Upon heating, the reaction mixture became very dark opaque brown, eventually turning black after 10 min at 40 °C. At the end of 2 h, the reaction mixture appeared to be opaque dark brown, perhaps not as black. The reaction mixture was then quenched by the addition of dimethyl disulfide (45 µL, 0.50 mmol), rapidly turning clear light yellow. The quenched reaction mixture was then stirred at 40 °C under nitrogen for 3 h. The solvent was evaporated to give a dark reddish solid. TLC indicated multiple streaked spots around the baseline that were of significantly lower Rf compared to the starting resorcinarene. 1H NMR showed the resorcinarene signals as broad humps, with multiple methoxy and aromatic signals, suggesting decomposition of the resorcinarene.
Attempted selective distal lithiation of 14,36,56,76-tetramethoxy-16,34,54,74-tetramethoxymethyl-2,4,6,8-tetrapropylresorcin[4]arene
The synthesis was performed based on the procedure by Arnott et al. [8]. To a clear colourless solution of resorcinarene methoxymethyl ether (3) (0.0497 g, 0.0559 mmol) in anhydrous THF (5 mL) at 40 °C was added dropwise n-butyllithium (0.18 mL, 1.6 M, 0.288 mmol). The clear slightly yellow solution rapidly turned clear dark yellow. The reaction mixture was allowed to stir at 40 °C under nitrogen for 2 h, appearing to be the same throughout. The reaction mixture was quenched by the addition of dimethyldisulfide (0.053 mL, 0.589 mmol), and was stirred under nitrogen, at 40 °C for 20 min. The THF solvent was then removed under reduced pressure to give a slightly yellow solid, which appeared as a mixture of resorcinarenes by 1H NMR spectroscopy, and of at least five compounds by TLC. The synthesis was repeated at the same scale (0.046 g, 0.052 mmol) with double the amount of n-butyllithium (0.33 mL, 1.6 M, 0.528 mmol, 10 equiv), giving a similar crude product with a sixth spot clearly visible by TLC. The two crude products were combined (0.093 g) and subjected to preparative TLC (EtOAc–petroleum spirits 40:60). The compounds were sufficiently separated to be clearly identified by 1H NMR spectroscopy as: tetra-SCH3 (8) (0.001 g, 0.9%), tri-SCH3 (7) (0.004 g, 3.6%), proximal-SCH3 (6) (0.009 g, 8.1%), distal-SCH3 (5) (0.005 g, 4.4%), mono-SCH3 (4) (0.028 g, 27.5%), and starting resorcinarene (1) (0.014 g, 14.6%).

Tetra-SCH3 resorcinarene (8): 1H NMR (only relevant signals quoted, CDCl3) δ 0.92 (t, J = 7.3 Hz, 12 H, CH2CH3), 1.27–1.38 (m, 8 H, CH2CH3), 1.76–1.91 (m, 8 H, CH2CH), 2.34 (s, 12 H, SCH3), 3.55, 3.64 (2s, 2 × 12 H, OCH 3 ), 4.63 (t, J = 7.5 Hz, 4 H, CHCH2), 4.89, 5.04 (2 × AB, J = 4.8 Hz, 8 H, OCH2O), 6.68 (s, 4 H, ArH).

Tri-SCH3 resorcinarene (7): 1H NMR (only relevant signals quoted, CDCl3) δ 0.77–1.02 (m, 12 H, CH2CH3), 1.19–1.48 (m, 8 H, CH2CH3), 1.59–1.97 (m, 8 H, CH2CH), 2.28, 2.43 (2s, 3 H, 6 H, SCH3), 3.25, 3.36, 3.45, 3.54, 3.64, 3.65, 3.80, 3.85 (8s, 8 × 3 H, OCH 3 ), 4.20, 4.63 (2 × AB, J = 4.7 Hz, OCH2O), 4.46–4.56, 4.54–4.62 (m, 2 × 2 H, CHCH2), 4.85, 4.90 (2 × AB, J = 6.7 Hz, 2 H, OCH2O), 5.00 (d, J = 4.7 Hz, 1 H, OCH2O), 5.05 (d, J = 4.7 Hz, 1 H, OCH2O), 5.17 (d, J = 4.7 Hz, 1 H, OCH2O), 5.19 (d, J = 4.7 Hz, 1 H, OCH2O), 6.35, 6.38, 6.42, 6.93, 6.95 (5s, 5 × 1 H, ArH).

Proximal-SCH3 resorcinarene (6): 1H NMR (only relevant signals quoted, CDCl3) δ 0.87–0.98 (m, 12 H, CH2CH3), 1.23–1.45 (m, 8 H, CH2CH3), 1.73–1.92 (m, 8 H, CH2CH), 2.32, 2.39 (2s, 2 × 3 H, SCH3), 3.31, 3.44, 3.49, 3.58, 3.65, 3.69 (6s, 3 H, 3 H, 6 H, 6 H, 3 H, 3 H, OCH 3 ), 4.37 (d, J = 4.4 Hz, 1 H, OCH2O), 4.45–4.61 (m, 4 H, CHCH2), 4.70 (d, J = 5.0 Hz, 1 H, OCH2O), 4.85 (d, J = 4.5 Hz, 1 H, OCH2O), 4.90–4.99 (m, 3 H, OCH2O), 5.04 (s, 2 H, OCH2O), 6.46, 6.49, 6.53, 6.55, 6.77, 6.78 (6s, 6 × 1 H, ArH).

Distal-SCH3 resorcinarene (5): 1H NMR (only relevant signals quoted, CDCl3) δ 0.91 (t, J = 6.7 Hz, 6 H, CH2CH3), 0.94 (t, J = 6.8 Hz, 6 H, CH2CH3), 1.19–1.50 (m, 8 H, CH2CH3), 1.72–1.97 (m, 8 H, CH2CH), 2.42 (s, 6 H, SCH3), 3.18, 3.55, 3.65, 3.80 (4s, 4 × 6 H, OCH 3 ), 4.45–4.56 (m, 4 H, CHCH2), 4.58, 4.73 (2 × AB, J = 6.7 Hz, 4 H, OCH2O), 4.96, 5.13 (2 × AB, J = 4.7 Hz, 4 H, OCH2O), 6.38, 6.44, 6.94 (3s, 3 × 2 H);

Mono-SCH3 resorcinarene (4): 1H NMR (only relevant signals quoted, CDCl3) δ 0.85–1.00 (m, 12 H, CH2CH3), 1.27–1.49 (m, 8 H, CH2CH3), 1.72–1.93 (m, 8 H, CH2CH), 2.37 (s, 3 H, SCH3), 3.29, 3.32, 3.40, 3.54, 3.59, 3.62, 3.63, 3.65 (8s, 8 × 3 H, OCH 3 ), 4.46–4.59 (m, 5 H, CHCH2 + OCH2O), 4.71 (d, J = 6.4 Hz, 1 H, OCH2O), 4.76 (d, J = 6.4 Hz, 1 H, OCH2O), 4.83 (d, J = 6.4 Hz, 1 H, OCH2O), 4.87 (d, J = 6.4 Hz, 1 H, OCH2O), 4.94 (d, J = 6.4 Hz, 1 H, OCH2O), 4.96 (d, J = 6.4 Hz, 1 H, OCH2O), 4.99 (d, J = 6.4 Hz, 1 H, OCH2O), 6.48, 6.49, 6.51, 6.59, 6.63, 6.69, 6.70 (7s, 7 × 1 H, ArH).
14,36,56,76-Tetraethoxy-16,34,54,74-tetramethoxy-15,55-di(methylthio)-2,4,6,8-tetrapropylresorcin[4]arene (9)

Synthesis was performed according to the procedure by Arnott et al. [8]. To a clear colourless solution of resorcinarene ethyl ether (10) (0.050 g, 0.061 mmol) in anhydrous THF (5 mL) at 40 °C was added dropwise n-butyllithium (0.19 mL, 1.6 M, 0.30 mmol). The clear colourless solution rapidly turned yellow. The reaction mixture was allowed to stir at 40 °C under nitrogen for 2 h, becoming almost clear colourless after 1 h. The clear colourless reaction mixture was quenched by the addition of dimethyldisulfide (55 µL, 0.61 mmol), and was stirred under nitrogen, at 40 °C for 20 min. The THF solvent was removed under reduced pressure from the reaction mixture to give the crude product as a white to off-white solid which turned light-brownish yellow on prolonged exposure to air. The crude product was subjected to column chromatography (EtOAc–petroleum spirits 10:90–20:80) to afford pure (9) as a clear colourless glassy solid (0.020 g, 36%): mp 155 °C (CHCl3/MeOH); 1H NMR (CDCl3) δ 0.86–0.97 (m, 12 H, CH2CH3), 1.05 (t, J = 7.0 Hz, 6 H, OCH2CH3), 1.21–1.52 (m, 8 H, CH2CH2CH3), 1.36 (t, J = 7.0 Hz, 6 H, OCH2CH3), 1.65–1.94 (m, 8 H, CHCH2), 2.42 (s, 6 H, SCH3), 3.43–3.58 (m, 2 H, OCH2), 3.51 (s, 6 H, OCH3), 3.71–3.83 (m, 2 H, OCH2), 3.76 (s, 6 H, OCH3), 3.83–3.95, 4.02–4.14 (2 m, 2 × 2 H, OCH2), 4.41–4.56 (m, 4 H, CHCH2), 6.22, 6.36, 6.87 (s, 3 × 2H, ArH); 13C NMR (CDCl3) δ 14.35, 14.37, 15.0, 16.0 (CH2CH3), 18.3 (SCH3), 21.51, 21.54 (CH2CH3), 36.66, 36.69 (CHCH2), 37.6, 37.8 (CH2CH), 55.3, 60.7 (OCH3), 64.4, 68.8 (OCH2), 97.4 (CH, Ar), 123.0, 124.2, 124.4 (C, Ar), 125.9, 126.6 (CH, Ar), 135.6, 135.8, 155.8, 156.0, 156.3, 156.9 (C, Ar). Found: C, 70.75; H, 8.41; C54H76O8S2; requires C, 70.70; H, 8.35%.
References
McIldowie, M.J., Mocerino, M., Skelton, B.W., White, A.H.: Facile lewis acid catalyzed synthesis of C4 symmetric resorcinarenes. Org. Lett. 2(24), 3869–3871 (2000). https://doi.org/10.1021/ol006608u
McIldowie, M.J., Mocerino, M., Ogden, M.I.: A brief review of Cn-symmetric calixarenes and resorcinarenes. Supramol. Chem. 22(1), 13–39 (2010). https://doi.org/10.1080/10610270902980663
Casnati, A., Pochini, A., Ungaro, R., Ugozzoli, F., Arnaud, F., Fanni, S., Schwing, M.-J., Egberink, R.J.M., de Jong, F., Reinhoudt, D.N.: Synthesis, complexation, and membrane transport studies of 1,3-alternate calix[4]arene-crown-6 conformers: a new class of cesium selective ionophores. J. Am. Chem. Soc. 117(10), 2767–2777 (1995). https://doi.org/10.1021/ja00115a012
Larsen, M., Jørgensen, M.: Selective halogen–lithium exchange reaction of bromine-substituted 25,26,27,28-tetrapropoxycalix[4]arene. J. Org. Chem. 61(19), 6651–6655 (1996). https://doi.org/10.1021/jo9609440
Van Loon, J.D., Arduini, A., Coppi, L., Verboom, W., Pochini, A., Ungaro, R., Harkema, S., Reinhoudt, D.N.: Selective functionalization of calix[4]arenes at the upper rim. J. Org. Chem. 55(21), 5639–5646 (1990). https://doi.org/10.1021/jo00308a024
Shivanyuk, A., Paulus, E.F., Böhmer, V., Vogt, W.: Selective derivatizations of resorcarenes. 4. General methods for the synthesis of C2v-symmetrical derivatives. J. Org. Chem. 63(19), 6448–6449 (1998). https://doi.org/10.1021/jo981386n
Lukin, O., Shivanyuk, A., Pirozhenko, V.V., Tsymbal, I.F., Kalchenko, V.I.: Synthesis, conformation, and binding properties of resorcarene tetrasulfonates. Asymmetric reorganization of pendant sulfonyl groups via intramolecular SO–H–O hydrogen bonds. J. Org. Chem. 63(25), 9510–9516 (1998). https://doi.org/10.1021/jo981751a
Ngodwana, L., Kleinhans, D.J., Smuts, A.-J., van Otterlo, W.A.L., Arnott, G.E.: Selective derivatisation of resorcinarene ethers via an ortholithiation approach. RSC Adv. 3(12), 3873–3876 (2013). https://doi.org/10.1039/C3RA00141E
Barrett, E.S., Irwin, J.L., Turner, P., Sherburn, M.S.: Efficient distal-difunctionalization of cavitand bowls. J. Org. Chem. 66(24), 8227–8229 (2001). https://doi.org/10.1021/jo015988+
Snieckus, V.: Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev. 90(6), 879–933 (1990). https://doi.org/10.1021/cr00104a001
Townsend, C.A., Bloom, L.M.: Studies of methoxymethyl-directed metalation. Tetrahedron Lett. 22(40), 3923–3924 (1981). https://doi.org/10.1016/S0040-4039(01)82027-X
Blair, A., Kazerouni, N.: Reactive chemicals and cancer. Cancer Causes Control 8(3), 473–490 (1997)
Buckley, B.R., Boxhall, J.Y., Page, P.C.B., Chan, Y., Elsegood, M.R.J., Heaney, H., Holmes, K.E., McIldowie, M.J., McKee, V., McGrath, M.J., Mocerino, M., Poulton, A.M., Sampler, E.P., Skelton, B.W., White, A.H.: Mannich and O-alkylation reactions of tetraalkoxyresorcin[4]arenes—the use of some products in ligand-assisted reactions. Eur. J. Org. Chem. 2006(22), 5117–5134 (2006). https://doi.org/10.1002/ejoc.200600590
Acknowledgements
We are grateful for support from the Australian Government Research Training Program Scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there are no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tan, D.A., Mocerino, M. Distal functionalisation of C4 symmetric tetramethoxyresorcinarene by selective lithiation. J Incl Phenom Macrocycl Chem 91, 71–80 (2018). https://doi.org/10.1007/s10847-018-0802-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-018-0802-4