
Abstract
To determine the effect of the preparation method on the physicochemical properties, such as crystallinity, particle size, solubility and dissolution, of the poorly water-soluble fenofibrate, different samples were prepared from fenofibrate and hydroxypropyl-β-cyclodextrin (HP-β-CD) in a 1:1 molar ratio, by physical mixing, trituration, kneading, solvent-evaporation and spray-drying techniques. The corresponding aqueous solubilities and dissolutions were studied in comparison with that of the drug powder. Characterization of these samples was accomplished using particle size determination, powder x-ray diffraction, differential scanning calorimetry, thermogravimetry, scanning electron microscopy and Fourier transform infrared spectroscopy. All preparations improved the solubility and dissolution of the drug, as compared with that of the drug powder (P < 0.05), in the following descending order of techniques utilized: spray-drying > solvent-evaporation > kneading > trituration > physical mixing. In particular, the aqueous solubility of fenofibrate from the spray-dried preparation was 85.93 ± 6.82 µg/ml, and the dissolution was about 90 % within 20 min. The drug was present in the crystalline state in the physically mixed, triturated and kneaded preparations. However, it was converted into the amorphous state in the solvent-evaporated and spray-dried preparations. The spray-dried preparation having the smallest particle size of 2.44 ± 0.03 μm gave discrete particles. Thus, in the development of a preparation of the poorly water-soluble fenofibrate with HP-β-CD, the preparation method exhibits a significant effect on the physicochemical properties of the drug. Among the various preparations tested in this study, the spray-dried sample, which provided the highest solubility and dissolution of fenofibrate, is strongly recommended for possible administration via the oral route.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fenofibrate is clinically used to normalize elevated blood cholesterol and triglycerides [1]. It is a very hydrophobic drug (log P = 5.24) [2] which is practically insoluble in water [3, 4]. The chemical entities exhibiting aqueous solubility <0.1 mg/ml are poorly absorbed from the gastrointestinal tract [5]. The BCS Class II includes drugs which exhibit impaired solubility in aqueous media [6], and fenofibrate is also placed in this class. Numerous techniques have been utilized to promote the aqueous solubility and dissolution of hydrophobic drugs, thereby enhancing their oral bioavailability [7–9]. Such processes encompass trituration, particle size reduction, micronization, use of surfactants, salt formation, complex formation, development of liposomes and solid dispersions [10–12]. Conversion of the crystalline forms into the amorphous states can remarkably improve the solubility and dissolution of hydrophobic drugs [11, 12]. This can be accomplished by loading the drug molecule in the inner lipophilic cavity of amorphous cyclodextrins, such as hydroxypropyl-β-cyclodextrin (HP-β-CD) [13–15]. Cyclodextrins are non-toxic compounds because of not being absorbed from the gastrointestinal tract [16]. The outer surface of a cyclodextrin molecule is hydrophilic whereas the central cavity is lipophilic [17]. Accordingly, lipophilic drug molecules can be housed in the central cavity where an inclusion complex is formed. Amongst cyclodextrins, HP-β-CD possesses greater ability to accommodate several lipophilic drug molecules [18]. During drug/cyclodextrin complex formation, no making or breaking of covalent linkages takes place [19, 20]. A number of weak intermolecular interactions between drug and cyclodextrin attach the lipophilic drug molecule to the cyclodextrin molecule [21–23]. The present work is a unique study focusing on the comparison of physicochemical properties of various fenofibrate/HP-β-CD preparations obtained via different preparation methods.
In the present research, aimed at determining the effect of different preparation methods on the physicochemical characteristics of the drug/HP-β-CD preparations, various samples were prepared from fenofibrate and HP-β-CD in 1:1 molar ratio [24] via physical mixing [25], trituration [26], kneading [27], solvent-evaporation [25] and spray-drying [27] techniques. Spray-drying is known to produce amorphous material due to rapid solvent evaporation [28]. The physicochemical properties were determined by solubility, dissolution, micromeritics measurements, powder x-ray diffraction (PXRD), differential scanning calorimetry (DSC), thermal gravimetric analysis (TGA), scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FT-IR).
Materials and methods
Materials
Fenofibrate and Hydroxypropyl-β-cyclodextrin (HP-β-CD) were supplied by Hanmi Pharm. Co. (Suwon, South Korea). All chemicals were of reagent grade.
Phase solubility study
The phase solubility study was performed in accordance with the Higuchi and Connors method [24]. Different concentrations of aqueous HP-β-CD solutions (0.312, 0.625, 1.25, 2.5, 5, 10, 20 and 40 mM) were prepared in water. An excess amount of fenofibrate was added to 5 ml of each of these solutions placed in a 15 ml conical tube. After vortex-mixing for 1 min, the tubes were agitated (220 rpm) in a water bath (25 °C) for 3 days. Then, the samples were filtered (0.45 μm). The filtrate (2 ml) was subjected to UV absorbance measurements at 295 nm using a UV visible spectrophotometer model Ultrospec 7000 (GE Healthcare Life Sciences; Uppsala, Sweden). The corresponding blank concentration of HP-β-CD (2 ml) was used as reference for each sample measured. The complexation efficiency (CE) and association constant (Kc) were calculated using Eq. (1) and (2), respectively.
where S0 is the intrinsic solubility. The slope was obtained by the straight line of the phase solubility graph.
Preparation of fenofibrate-loaded samples
Physical mixing
The physical blending of drug and cyclodextrin was achieved simply by mixing fenofibrate and HP-β-CD (1:1 molar ratio) for 5 min to obtain a homogenous mixture. Subsequently, this mixture was passed through the sieve no. 60.
Trituration/Co-grinding
The drug and cyclodextrin (1:1 molar ratio) were mechanically ground together for 20 min using pestle and mortar. The comprehensively triturated blend was then passed through the sieve no. 60.
Kneading
The HP-β-CD was impregnated with little quantity of 90 % ethanol. The drug was gradually added to this semi-solid paste of HP-β-CD and kneaded for 20 min using pestle and mortar. The thoroughly kneaded mixture was dried in an oven at 40 °C. The dried mixture was pulverized and sieved through the sieve no. 60.
Solvent evaporation
The two components were completely dissolved in 90 % ethanol in a beaker by magnetic stirring. The resultant clear solution was evaporated in an oven at 40 °C for 48 h. The dried residue was powdered and passed through sieve no. 60.
Spray drying
Fenofibrate and HP-β-CD (1:1 molar ratio) were completely dissolved in 95 % ethanol. The resulting clear solution was spray-dried using Büchi B-290 mini spray dryer (Labortechnik AG Co., Meierseggstrasse, Flawil, Switzerland). The solution was pushed (7 ml/min) to the pneumatic nozzle (0.7 mm I.D.) by a peristaltic pump. The spraying air flowed at 4 kg/cm2 pressure. The atomized droplets were dried by hot air-flow. The inlet temperature was fixed at 100 °C which resulted in 65 °C outlet temperature. The aspirator setting at 100 % indicated a filter vessel pressure at −50 mbar. The dried product was obtained in the collecting vessel.
Determination of drug content
A 1:1 fenofibrate/HP-β-CD preparation having 10 mg fenofibrate, was completely dissolved in 100 ml acetonitrile. Then, 1 ml solution was filtered through 0.45 μm disc filter. An aliquot of 50 μl filtrate was analyzed by the HPLC method as mentioned below. The test was performed in triplicate. The drug content (%) was calculated according to the following formula:
where “CD” is the drug content, “CA” is the actual concentration (μg/ml) detected by the HPLC and “CT” is the theoretical concentration (μg/ml).
Aqueous solubility
An excess amount of the drug powder or inclusion preparation (about 100 mg) was added to 2 ml distilled water in a conical tube. Following vortex-mixing for 1 min, each vortex-mixed sample tube was placed in a water bath at 25 °C and agitated at 100 rpm for 7 days. Then, centrifugation was performed at 10,000 g for 10 min (Hanil Science Industrial Co., Smart 15; Gangneung, South Korea). One milliliter of clear supernatant was filtered (0.45 μm) into the sample vial. An aliquot of 50 μl filtrate was analyzed by the HPLC system (Agilent 1260 Infinity, Agilent Technologies, Santa Clara, CA, USA). The HPLC system was equipped with Capcell Pak C18 column (Shiseido, 4.6 mm I.D. 250 mm×, 5 μm) at 30 °C. The mobile phase consisting of acetonitrile/0.1 % (v/v) aqueous phosphoric acid (75/25, volume ratio) was flowed at 2 ml/min. Fenofibrate detection was accomplished at 286 nm wavelength [29].
Dissolution
USP dissolution apparatus II (Vision® Classic 6™, Hanson Research Co., Los Angeles, CA, USA) was used for conducting the dissolution behavior of the drug. The drug powder or the corresponding preparation having 80 mg fenofibrate, was enclosed in a hard gelatin capsule and inserted into the sinker. Dissolution of poorly water-soluble or water-insoluble chemical substances needs special dissolution medium. For such chemical entities, use of only water as a dissolution medium is highly discouraged. Therefore, the FDA has suggested the use of a slight quantity of a surfactant in the dissolution medium [30]. Incorporation of a surface active agent in dissolution medium improves the efficiency of dissolution of such APIs. Therefore, the loaded sinker was immersed into 900 ml dissolution medium containing 2 % (w/v) Polysorbate 80 [31]. The temperature of the dissolution medium vessel was maintained at 37 ± 0.1 °C by the surrounding water-bath. The paddle rotation was 100 rpm. At predetermined time intervals, one milliliter sample of the dissolution medium was withdrawn, filtered (0.45 μm) and quantified for fenofibrate by the HPLC method described above.
Particle size determination
Particle size analysis was carried out using the HELOS laser diffraction particle size analyzer (Model H1918, Sympatec GmbH, System-Partikel-Technik, Clausthal-Zellerfeld, Germany). The cumulative undersize distribution was expressed as X10, X50 and X90 (diameters) at the undersize values of 10, 50 and 90 % of total volume shown by the cumulative undersize (%) curves, respectively.
Powder x-ray diffraction (PXRD)
The crystalline property of the samples was established using a Rigaku x-ray diffractometer (D/MAX-2500 PC, Rigaku Corporation; Tokyo, Japan). The instrument was equipped with a miniflex goniometer and a Cu Kα1 monochromatic radiation source. The analysis was executed in the range of 10–80 °C with a step size of 0.02°/second, scanning mode of 2θ and 5°/min scanning speed using 40 mA current and 40 kV voltage [32].
Differential scanning calorimetry (DSC)
For determining thermal characteristics, each sample (about 5 mg) sealed in aluminum pan was subjected to heating in the range of 30–130 °C at the rate of 10 °C/min using a nitrogen flow of 25 cm3/minute, using the differential scanning calorimeter (DSC Q20, TA Instruments; New Castle, Delaware, USA).
Thermogravimetric analysis (TGA)
TGA analysis was performed using the thermogravimetric analyzer (TGA Q50, TA Instruments; New Castle, Delaware, USA). The sample (2–10 mg) placed in a platinum pan was carried to the furnace by the moving arm. The weight loss (%) was determined while heating the sample in the range of 30–140 °C at the rate of 10 °C/min using a nitrogen flow of 25 cm3/min.
Scanning electron microscopy (SEM)
The shape and surface properties of the samples were inspected with a scanning electron microscope (S-4800, Hitachi; Tokyo, Japan). The double adhesive carbon tape was attached onto the sample loading disc. Then, the samples were affixed to the open surface of adhered tape. Subsequently, the samples were coated with platinum for 4 min to facilitate electrical transmission. For this purpose, EMI Teck Ion Sputter (K575X) was used. The coating was performed under 7 × 10−3 mbar pressure, 25 mA current and 100 % turbo speed.
Fourier transform infrared spectroscopy (FT-IR)
An FT-IR spectrophotometer (Nicolet-6700; Nicolet Instrument Corporation; Madison, Wisconsin, USA) was used to examine the IR spectra of different samples for possible drug cyclodextrin interactions. Each powder sample was directly placed on the sample disc and scanned in the range of 400–4,000 cm−1. The FT-IR spectra were obtained using the OMNIC software (Nicolet Instrument Corporation; Madison, Wisconsin, USA).
Results and discussion
Fenofibrate normalizes abnormally increased concentration of cholesterol and triglycerides in plasma [1]. It is a BCS Class II drug [6]. Accordingly, after oral administration it furnishes poor bioavailability due to insufficient absorption from the gastrointestinal tract [5]. In this study, HP-β-CD was used as a carrier because of its greater affinity for lipophilic molecules [18]. The outer surface of HP-β-CD is hydrophilic whereas the inner cavity is lipophilic [17]. Therefore, lipophilic fenofibrate can be accommodated into the central cavity of HP-β-CD, where an inclusion complex is formed [18].
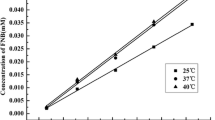
Before the preparation of a fenofibrate/HP-β-CD sample, a phase solubility study was performed in accordance with the Higuchi and Connors phase solubility study method [24]. The results of the phase solubility study are shown in Fig. 1. A straight line (r2 = 0.9999) was obtained in the phase solubility study. The stability constant (Kc) and complexation efficiency (CE) calculated from the slope of the line were 3.199 × 103 M−1 and 3.11 × 10−3, respectively. Accordingly, a 1:1 molar ratio of fenofibrate and HP-β-CD was opted [24] for the preparation of various samples via the physical mixing, trituration, kneading, solvent-evaporation and spray-drying techniques. The drug contents measured in the solid state fenofibrate/HP-β-CD samples prepared by the physical mixing, trituration, kneading, solvent-evaporation and spray-drying method were 85.49 ± 12.16, 89.12 ± 12.06, 90.26 ± 1.50, 98.07 ± 4.06 and 101.10 ± 5.64 %, respectively. In the solvent-evaporation and spray-drying methods, the constituents were completely dissolved in ethanol prior to evaporation. As a result, the drug content in these formulations was higher as compared to other formulations.

The fenofibrate/HP-β-CD phase solubility diagram obtained in water at 25 °C. Each value denotes the mean ± S.D. (n = 3)
The aqueous solubilities, at 25 °C, of the drug samples in the form of a fenofibrate powder, physically mixed, triturated, kneaded, solvent-evaporated, and spray-dried samples were found equivalent to 0.35 ± 0.05, 0.49 ± 0.30, 10.25 ± 1.06, 30.66 ± 2.61, 65.89 ± 4.74, and 85.93 ± 6.82 μg/ml, respectively (Fig. 2). In particular, the aqueous solubility of fenofibrate in the spray-dried sample was about 250 times that of the parent drug powder. Similarly, the mass percent dissolution, at 37 °C, of the drug from the fenofibrate powder, physically mixed, triturated, kneaded, solvent-evaporated, and spray-dried samples measured at 40 min were 22.88 ± 1.41, 30.11 ± 1.85, 69.02 ± 1.67, 78.52 ± 1.29, 82.22 ± 1.93 and 95.06 ± 0.30 %, respectively (Fig. 3). In particular, initial dissolution of the drug from the spray-dried sample was comparatively more rapid.

Effect of the 1:1 fenofibrate/HP-β-CD preparation method on the aqueous solubility of fenofibrate in water at 25 °C. Each value represents the mean ± S.D. (n = 3): a fenofibrate powder; b physically mixed preparation; c triturated preparation; d kneaded preparation; e solvent-evaporated preparation; f spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio. P < 0.05 for each preparation as compared to the drug powder

Effect of the 1:1 fenofibrate/HP-β-CD preparation method on the dissolution of fenofibrate in dissolution medium at 37 °C. Each value represents the mean ± S.D. (n = 6): a fenofibrate powder; b physically mixed preparation; c triturated preparation; d kneaded preparation; e solvent-evaporated preparation; f spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio.*P < 0.05 for each preparation as compared to the drug powder
The particle-size cumulative distribution data are listed in Table 1 and depicted in Fig. 4. The particle size distributions of the physically mixed, triturated, kneaded, solvent-evaporated, and spray-dried samples were recorded as X10, X50 and X90-values from the cumulative undersize (%) curves [33, 34]. The corresponding median particle size (X50) were 34.57 ± 2.81, 11.42 ± 0.08, 17.89 ± 4.08, 13.30 ± 0.78 and 2.44 ± 0.03 μm, respectively. In particular, the mean particle surface area of the spray-dried sample was considerably great (32,105.43 ± 5,021.32 cm2/g). Moreover, the physically mixed (Fig. 4a), triturated (Fig. 4b), kneaded (Fig. 4c) and solvent-evaporated (Fig. 4d) samples exhibited wide particle-size distributions. In contrast, the particle-size distribution proved narrow in case of the spray-dried sample (Fig. 4e). As compared to the physically mixed sample, the median particle sizes (X50) of other samples were significantly lower (P < 0.05). The median particle-size (X50) of the triturated sample was reduced due to vigorous grinding and prolonged mixing of constituents for 20 min. The median particle-size (X50) of the kneaded sample was not very different from that of the solvent-evaporated sample. Their particle sizes were larger than that of the triturated sample because of a relatively short time of grinding (1–2 min) during pulverization of the dried residue. However, the median particle-size (X50) of the spray-dried sample was significantly reduced (P < 0.05) in comparison with other samples. The reduced particle size led to enhancement of the surface area available for dissolution and, hence, the dissolution rate was improved [35].

Particle size distribution for a physically mixed preparation; b triturated preparation; c kneaded preparation; d solvent-evaporated preparation; e spray-dried preparation: Cumulative undersize distribution (%), the particle sizes at the undersize values (mean ± S.D., n = 3) of 10 % (X10), 50 % (X50) and 90 % (X90). X50 is the median particle size
Figure 5 reveals the structural aspects of samples. Fenofibrate powder generated sharp characteristic peaks confirming the crystalline nature of the drug (Fig. 5a). HP-β-CD did not produce any peaks suggesting the amorphous nature of the hydrophilic cyclic polymer (Fig. 5b). The distinctive peaks associated with fenofibrate crystalline form also appeared in the PXRD patterns of the physically mixed (Fig. 5c), triturated (Fig. 5d) and kneaded (Fig. 5e) preparations. This suggested the presence of the crystalline drug in these samples. However, no peak was seen in the PXRD patterns of the solvent-evaporated (Fig. 5f) and spray-dried (Fig. 5g) samples. This confirmed that the drug was converted into the amorphous state after inclusion into the HP-β-CD cavity in both the solvent-evaporation and spray-dying techniques.

PXRD patterns: a fenofibrate powder; b HP-β-CD; c physically mixed preparation; d triturated preparation; e kneaded preparation; f solvent-evaporated preparation; g spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio
Similarly, the DSC results (Fig. 6) show that the fenofibrate powder (Fig. 6a) produced a deep endotherm corresponding to its melting point at about 81.5 °C showing its typical crystalline nature. The HP-β-CD showed no sharp endotherm confirming its amorphous state (Fig. 6b). However, a broad descending curve was due to loss of water bound with the HP-β-CD [36]. Furthermore, the sharp endotherm corresponding to the melting point of crystalline fenofibrate was also witnessed in case of the physically mixed (Fig. 6c), triturated (Fig. 6d) and kneaded (Fig. 6e) samples. However, no sharp endotherm appeared for the solvent-evaporated (Fig. 6f) and spray-dried (Fig. 6g) samples. Like PXRD results, DSC results also confirmed that the drug was present in the crystalline state in the physically mixed, triturated and kneaded samples. On the other hand, it was a clear change to the amorphous form in the solvent-evaporated and spray-dried samples. Amorphous drugs are markedly more soluble than their corresponding crystalline forms [33, 37]. However, recrystallization might be expected at supersaturation. The HP-β-CD improved aqueous solubility of the drug by inhibiting recrystallization [38–40].

DSC thermograms: a fenofibrate powder; b HP-β-CD; c physically mixed preparation; d triturated preparation; e kneaded preparation; f solvent-evaporated preparation; g spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio
Figure 7 shows the results of thermogravimetric analysis in the range of 30–140 °C. In the case of fenofibrate powder, a straight line was obtained (Fig. 7a). The HP-β-CD produced a broad descending curve in the range of 30–100 °C (Fig. 7b). This curve indicated the loss of bound water in this temperature range [36]. Likewise, this behavior was also observed in case of the physically mixed (Fig. 7c), triturated (Fig. 7d), kneaded (Fig. 7e), solvent-evaporated (Fig. 7f) and spray-dried (Fig. 7g) samples. Moreover, the weight losses of fenofibrate powder, HP-β-CD, physically mixed, triturated, kneaded, solvent-evaporated and spray-dried samples were 0.27, 4.89, 3.09, 3.06, 3.53, 3.14 and 3.21 %, respectively. The results showed that the losses of weight in case of the physically mixed, triturated, kneaded, solvent-evaporated and spray-dried samples were not very much different from one another. Furthermore, the water of hydration was strongly bound to HP-β-CD, and did not completely evaporate during drying processes used in the preparation of solvent-evaporated and spray-dried samples [36].

TGA thermograms: a fenofibrate powder; b HP-β-CD; c physically mixed preparation; d triturated preparation; e kneaded preparation; f solvent-evaporated preparation; g spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio
The shape and surface features of fenofibrate powder, physically mixed, triturated, kneaded, solvent-evaporated and spray-dried samples are shown in Fig. 8. The scanning electron micrograph of fenofibrate powder showed asymmetrical crystals (Fig. 8a). The HP-β-CD contained various-sized larger particles. Therefore, the SEM images of HP-β-CD (Fig. 8b) and the physically mixed product (Fig. 8c) were taken at lower magnification. Similarly, the micrographs of the triturated (Fig. 8d), kneaded (Fig. 8e) and solvent-evaporated (Fig. 8f) samples also showed different-sized particles with irregular shapes and surfaces. However, the discrete particles of the spray-dried sample were of regular shape and size, but irregular surface (Fig. 8g).

SEM images: a fenofibrate powder (×2000); b HP-β-CD (×200); c physically mixed preparation (×200); d triturated preparation (×2000); e kneaded preparation (×2000); f solvent-evaporated preparation (×2000); g spray-dried preparation (×2000). Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio
The FT-IR spectroscopic results are displayed in Fig. 9. The distinguishing peaks of fenofibrate were appeared at 923, 1,597, 1,648 and 1,726 cm−1 (Fig. 9a). These peaks were also observed in the spectra of the physically mixed, triturated, kneaded, solvent-evaporated and spray-dried samples. Moreover, the physically mixed (Fig. 9c), triturated (Fig. 9d), kneaded (Fig. 9e), solvent-evaporated (Fig. 9f) and spray-dried (Fig. 9g) samples exhibited similar spectral patterns. There was no apparent shift of peaks or emergence of new peaks, confirming the absence of making or breaking of new chemical bonds between the drug and cyclodextrin [19, 20, 41].

FT-IR spectrograms: a fenofibrate powder; b HP-β-CD; c physically mixed preparation; d triturated preparation; e kneaded preparation; f solvent-evaporated preparation; g spray-dried preparation. Each preparation contained fenofibrate and HP-β-CD in a 1:1 molar ratio
As compared with the fenofibrate powder, the physically mixed sample improved the aqueous solubility and dissolution of the drug due to the presence of the hydrophilic polymer, which enhanced wettability [42, 43]. The solubility and dissolution of the drug from the triturated sample was better than that of the physically mixed sample because of decrease in particle size [44, 45]. Irrespective of a relatively larger particle size, the solubility and dissolution of the drug from the kneaded sample was better than that from the triturated sample. In the preparation of the kneaded sample, the hydrophilic polymer was impregnated with ethanol first, then the drug was let to gradually mixed with HP-β-CD on continuous kneading. Accordingly, the enhanced solubility and dissolution can be ascribed to comparatively closer contact between the drug and the hydrophilic polymer in the kneaded sample [33]. The PXRD and DSC results showed that the drug was present in the amorphous form in the solvent-evaporated and spray-dried samples. The solubility of the drug with the solvent-evaporated sample was greater than those of the triturated and kneaded samples. This enhanced solubility was due to the amorphous state of the drug [12]. However, the initial dissolution rate of the solvent-evaporated sample was not significantly different (P > 0.05) from those of the triturated and the kneaded samples. This might be due to their similar particle-size distributions (P < 0.05). As particles dissolve slowly, the loaded drug would also dissolve slowly [46]. The spray-dried sample demonstrated the highest solubility and dissolution of the drug owing to a combination of factors including the conversion into the amorphous state [47], reduction in particle size with a narrow size distribution [45], enhanced surface area available for dissolution [35] and improved wetting due to more homogeneity of the constituents and closeness of the hydrophilic polymer with the amorphous drug [11, 42, 43].
Thus, all sample preparation methods improved the solubility and dissolution of the drug in comparison with the drug powder (P < 0.05) in the decreasing order of spray-dried > solvent-evaporated > kneaded > triturated > physically mixed samples. The drug was in the crystalline state in the physically mixed, triturated and kneaded samples. However, it was changed into the amorphous form in the solvent-evaporated and spray-dried samples. In particular, the discrete microparticles (X50 = 2.44 ± 0.03 μm) of the spray-dried sample containing the amorphous drug (drug content = 101.10 ± 5.64 %) exhibited the highest drug solubility (85.93 ± 6.83 μg/ml) and dissolution (about 80 % within 10 min). Thus, the fenofibrate/HP-β-CD inclusion sample (1:1 molar ratio) prepared by the spray-drying technique could be used to remarkably enhance the aqueous solubility and dissolution of poorly water-soluble fenofibrate.
Conclusion
All preparations exhibited better aqueous drug solubility and dissolution than that of the drug powder. The enhanced drug dissolution rate can be attributed to (i) an improved solubility of the drug as a result of its amorphous state or small particle size (Kelvin’s law) [44, 45, 47], (ii) an increased surface area available for drug dissolution because of a relatively smaller size of the drug particle [48, 49], (iii) an improved wetting of the drug by the hydrophilic carrier [42, 43] and (iv) the presence of hydrophobic methyl groups which extend the hydrophobic cavity of the cyclodextrin molecule [41]. This work emphasized that in the development of an inclusion preparation of the poorly water-soluble drugs with HP-β-CD, the preparation method plays profound role on the drug physicochemical characteristics, such as crystallinity, particle size, solubility and dissolution. Among the various preparations examined in this work, including physical mixing, trituration, kneading, solvent-evaporation, spray-drying, the latter technique of spray-drying a 1:1 fenofibrate and HP-β-CD solution of fenofibrate/HP-β-CD in 95 % aqueous ethanol exhibited the best enhanced drug solubility and dissolution, and is thus strongly recommended for enhanced fenofibrate delivery.
References
Balfour, J.A., McTavish, D., Heel, R.C.: Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs 40(2), 260–290 (1990)
Munoz, A., Guichard, J.P., Reginault, P.: Micronised fenofibrate. Atherosclerosis 110, Suppl(0), S45–S48 (1994). doi:http://dx.doi.org/10.1016/0021-9150(94)05375-S
Ming-Thau, S., Ching-Min, Y., Sokoloski, T.D.: Characterization and dissolution of fenofibrate solid dispersion systems. Int. J. Pharm. 103(2), 137–146 (1994). doi:http://dx.doi.org/10.1016/0378-5173(94)90094-9
Vogt, M., Kunath, K., Dressman, J.B.: Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: comparison with commercial preparations. Eur. J. Pharm. Biopharm. 68(2), 283–288 (2008)
Dressman, J., Butler, J., Hempenstall, J., Reppas, C.: The BCS: where do we go from here? Pharm. Technol. 25(7), 68–77 (2001)
Amidon, G., Lennernäs, H., Shah, V., Crison, J.: A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12(3), 413–420 (1995). doi:10.1023/a:1016212804288
Badens, E., Majerik, V., Horváth, G., Szokonya, L., Bosc, N., Teillaud, E., Charbit, G.: Comparison of solid dispersions produced by supercritical antisolvent and spray-freezing technologies. Int. J. Pharm. 377(1–2), 25–34 (2009). doi:http://dx.doi.org/10.1016/j.ijpharm.2009.04.047
Cutler, L., Howes, C., Deeks, N.J., Buck, T.L., Jeffrey, P.: Development of a P-glycoprotein knockout model in rodents to define species differences in its functional effect at the blood–brain barrier. J. Pharm. Sci. 95(9), 1944–1953 (2006)
Yoshihashi, Y., Iijima, H., Yonemochi, E., Terada, K.: Estimation of physical stability of amorphous solid dispersion using differential scanning calorimetry. J. Therm. Anal. Calorim. 85(3), 689–692 (2006). doi:10.1007/s10973-006-7653-8
Goddeeris, C., Coacci, J., Van den Mooter, G.: Correlation between digestion of the lipid phase of smedds and release of the anti-HIV drug UC 781 and the anti-mycotic drug enilconazole from smedds. Eur. J. Pharm. Biopharm. 66(2), 173–181 (2007). doi:http://dx.doi.org/10.1016/j.ejpb.2006.10.005
Leuner, C., Dressman, J.: Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 50(1), 47–60 (2000). doi:http://dx.doi.org/10.1016/S0939-6411(00)00076-X
Perrut, M., Jung, J., Leboeuf, F.: Enhancement of dissolution rate of poorly-soluble active ingredients by supercritical fluid processes: Part I: micronization of neat particles. Int. J. Pharm. 288(1), 3–10 (2005). doi:http://dx.doi.org/10.1016/j.ijpharm.2004.09.007
Antoniadou-Vyza, E., Buckton, G., Michaleas, S.G., Loukas, Y.L., Efentakis, M.: The formation of an inclusion complex of methocarbamol with hydroxypropyl-β-cyclodextrin: the effect on chemical stability, solubility and dissolution rate. Int. J. Pharm. 158(2), 233–239 (1997). doi:http://dx.doi.org/10.1016/S0378-5173(97)00258-5
Müller, B.W., Brauns, U.: Solubilization of drugs by modified β-cyclodextrins. Int. J. Pharm. 26(1–2), 77–88 (1985). doi:http://dx.doi.org/10.1016/0378-5173(85)90201-7
Pitha, J., Pitha, J.: Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J. Pharm. Sci. 74(9), 987–990 (1985). doi:10.1002/jps.2600740916
Irie, T., Uekama, K.: Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J. Pharm. Sci. 86(2), 147–162 (1997). doi:10.1021/js960213f
Loftsson, T., Jarho, P., Masson, M., Järvinen, T.: Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2(2), 335–351 (2005)
Miro, A., Quaglia, F., Giannini, L., Cappello, B., Giannini, L., Immacolata La Rotonda, M.: Drug/cyclodextrin solid systems in the design of hydrophilic matrices: a strategy to modulate drug delivery rate. Curr. Drug Deliv. 3(4), 373–378 (2006). doi:10.2174/156720106778558994
Loftsson, T., Brewster, M.E.: Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 85(10), 1017–1025 (1996)
Thompson, D.O.: Cyclodextrins— enabling excipients: their present and future use in pharmaceuticals. 14(1), 104 (1997). doi:10.1615/CritRevTherDrugCarrierSyst.v14.i1.10
Connors, K.A.: The stability of cyclodextrin complexes in solution. Chem. Rev. 97(5), 1325–1358 (1997)
Li, S., Purdy, W.C.: Cyclodextrins and their applications in analytical chemistry. Chem. Rev. 92(6), 1457–1470 (1992)
Rekharsky, M.V., Inoue, Y.: Complexation thermodynamics of cyclodextrins. Chem. Rev. 98(5), 1875–1918 (1998)
Higuchi, T., Connors, K.A.: Phase-solubility techniques. Adv. Anal. Chem. Instrum. 4(2), 117–212 (1965)
Patil, J., Kadam, D., Marapur, S., Kamalapur, M.: Inclusion complex system; a novel technique to improve the solubility and bioavailability of poorly soluble drugs: a review. Int. J. Pharm. Sci. Rev. Res. 2(2), 29–33 (2010)
Astakhova, A.V., Demina, N.B.: Modern drug technologies: synthesis, characterization, and use of inclusion complexes between drugs and cyclodextrins (a review). Pharm. Chem. J. 38(2), 105–108 (2004). doi:10.1023/B:PHAC.0000032490.04705.ba
Fernandes, C.M., Veiga, F.J.B.: Effect of the hydrophobic nature of triacetyl-beta-cyclodextrin on the complexation with nicardipine hydrochloride: physicochemical and dissolution properties of the kneaded and spray-dried complexes. Chem. Pharm. Bull. 50(12), 1597–1602 (2002). Tokyo
Sebhatu, T., Angberg, M., Ahlneck, C.: Assessment of the degree of disorder in crystalline solids by isothermal microcalorimetry. Int. J. Pharm. 104(2), 135–144 (1994)
Mehmood Yousaf, A., Wuk Kim, D., Choi, H.-G., Oh, E.: Validation of a highly sensitive RP-HPLC method for quantification of fenofibrate in pure and pharmaceutical dosage forms. Curr. Pharm. Anal. 10(2), 97–104 (2014)
Shah, V.P., Noory, A., Noory, C., McCullough, B., Clarke, S., Everett, R., Naviasky, H., Srinivasan, B., Fortman, D., Skelly, J.P.: In vitro dissolution of sparingly water-soluble drug dosage forms. Int. J. Pharm. 125(1), 99–106 (1995)
Stamm, A., Seth, P.: Fenofibrate pharmaceutical composition having high bioavailabilty. In. Google Patents, (2006). http://www.google.com/patents/US6277405
Sanganwar, G.P., Gupta, R.B.: Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int. J. Pharm. 360(1–2), 213–218 (2008). doi:http://dx.doi.org/10.1016/j.ijpharm.2008.04.041
Joe, J.H., Lee, W.M., Park, Y.-J., Joe, K.H., Oh, D.H., Seo, Y.G., Woo, J.S., Yong, C.S., Choi, H.-G.: Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int. J. Pharm. 395(1–2), 161–166 (2010). doi:http://dx.doi.org/10.1016/j.ijpharm.2010.05.023
Konno, H., Handa, T., Alonzo, D.E., Taylor, L.S.: Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur. J. Pharm. Biopharm. 70(2), 493–499 (2008)
Noyes, A.A., Whitney, W.R.: The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 19(12), 930–934 (1897)
Mihajlovic, T., Kachrimanis, K., Graovac, A., Djuric, Z., Ibric, S.: Improvement of aripiprazole solubility by complexation with (2-hydroxy) propyl-β-cyclodextrin using spray drying technique. AAPS Pharm. Sci. Tech. 13(2), 623–631 (2012)
Hancock, B.C., Parks, M.: What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 17(4), 397–404 (2000)
Dias, M.M.R., Raghavan, S.L., Pellett, M.A., Hadgraft, J.: The effect of β-cyclodextrins on the permeation of diclofenac from supersaturated solutions. Int. J. Pharm. 263(1–2), 173–181 (2003). doi:http://dx.doi.org/10.1016/S0378-5173(03)00366-1
Derakhshanian, V., Banerjee, S.: Cyclodextrin inhibits calcium carbonate crystallization and scaling. Ind. Eng. Chem. Res. 51(11), 4463–4465 (2012)
Clas, S.D., Kwong, E., Meisner, D., Vadas, E.B., Winters, C.: Topical polymeric drug delivery system. In. Google Patents, (1997). http://www.google.com/patents/EP0758229A1?cl=en
Upadhye, S.B., Kulkarni, S.J., Majumdar, S., Avery, M.A., Gul, W., ElSohly, M.A., Repka, M.A.: Preparation and characterization of inclusion complexes of a hemisuccinate ester prodrug of Δ9-tetrahydrocannabinol with modified beta-cyclodextrins. AAPS Pharm. Sci. Tech. 11(2), 509–517 (2010)
Chow, A.H., Hsia, C.K., Gordon, J.D., Young, J.W., Vargha-Butler, E.I.: Assessment of wettability and its relationship to the intrinsic dissolution rate of doped phenytoin crystals. Int. J. Pharm. 126(1), 21–28 (1995)
Kawashima, Y., Saito, M., Takenaka, H.: Improvement of solubility and dissolution rate of poorly water-soluble salicylic acid by a spray-drying technique. J. Pharm. Pharmacol. 27(1), 1–5 (1975)
Dai, W.-G., Dong, L.C., Song, Y.-Q.: Nanosizing of a drug/carrageenan complex to increase solubility and dissolution rate. Int. J. Pharm. 342(1), 201–207 (2007)
Mura, P., Cirri, M., Faucci, M., Ginès-Dorado, J., Bettinetti, G.: Investigation of the effects of grinding and co-grinding on physicochemical properties of glisentide. J. Pharm. Biomed. Anal. 30(2), 227–237 (2002)
Srinarong, P., Faber, J., Visser, M., Hinrichs, W., Frijlink, H.: Strongly enhanced dissolution rate of fenofibrate solid dispersion tablets by incorporation of superdisintegrants. Eur. J. Pharm. Biopharm. 73(1), 154–161 (2009)
Yonemochi, E., Kitahara, S., Maeda, S., Yamamura, S., Oguchi, T., Yamamoto, K.: Physicochemical properties of amorphous clarithromycin obtained by grinding and spray drying. European J. Pharm. Sci. 7(4), 331–338 (1999)
Kubo, H., Mizobe, M.: Improvement of dissolution rate and oral bioavailability of a sparingly water-soluble drug, ()-5-[[2-(2-Naphthalenylmethyl)-5-benzoxazolyl] methyl]-2, 4-thiazolidinedione, in Co-ground Mixture with d-Mannitol. Biol. Pharm. Bull. 20, 460–463 (1997)
Purvis, T., Vaughn, J.M., Rogers, T.L., Chen, X., Overhoff, K.A., Sinswat, P., Hu, J., McConville, J.T., Johnston, K.P., Williams III, R.O.: Cryogenic liquids, nanoparticles, and microencapsulation. Int. J. Pharm. 324(1), 43–50 (2006)
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. NRF-2012R1A2A2A01045658).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Yousaf, A.M., Kim, D.W., Cho, K.H. et al. Effect of the preparation method on crystallinity, particle size, aqueous solubility and dissolution of different samples of the poorly water-soluble fenofibrate with HP-β-CD. J Incl Phenom Macrocycl Chem 81, 347–356 (2015). https://doi.org/10.1007/s10847-014-0461-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-014-0461-z