
Abstract
Recently, \(\hbox {Gd}_{2}\hbox {O}_{3}\) has gained considerable interest in industry, and its optical applications have been of interest in optoelectronic. The band structure and optical properties of cubic \(\hbox {Gd}_{2}\hbox {O}_{3}\) are investigated using the density functional theory framework. Calculations are performed within the local density approximation and generalized gradient approximation, adding the empirical Hubbard potential U. Calculation of the electronic band structure indicates a direct \({\Gamma }\) band gap. Further, the total and partial densities of states were presented, and the contribution of different orbitals is analyzed. Moreover, the behavior of optical spectra such as real and imaginary part of dielectric function, refractive index, extinction coefficient, optical conductivity, and electron energy-loss function is analyzed. There is a good agreement between the computed results and reported experimental data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Rare-earth sesquioxides (\(\hbox {RE}_{2}\hbox {O}_{3}\), RE = rare earth) find important applications in microelectronic, optoelectronic, and the optical device industry because of their unique structural and electronic properties [1], which result from their 4f electrons. They are used as additives for low-temperature sintering in ceramics; moreover, the sesquioxides are of interest as grain growth inhibitors and as phase stabilizers [2]. The structural flexibility of rare-earth sesquioxides motivates their applications in solid oxide fuel cells (SOFC) as oxygen-ion conductors, oxygen sensors, membrane reactors for oxidative catalysis, and dense ceramic membranes for oxygen separation [3].
At ambient conditions, rare-earth sesquioxides are found in three A, B, and C polymorphic forms. The A phase has the P-3m1 space group with a hexagonal structure, the B phase has the C2/m space group with a monoclinic structure, while the C phase with a cubic structure has the Ia-3 space group. For most of sesquioxides with medium size cations, at standard conditions the crystal structure can be either cubic or monoclinic. \(\hbox {Gd}_{2}\hbox {O}_{3}\) was usually observed in the cubic structure [4]. \(\hbox {Gd}_{2}\hbox {O}_{3}\) is a very promising and suitable material with a wide range of applications in various technological fields. It has been considered a protective material due to its thermal stability and corrosion resistivity [5]. The \(\hbox {Gd}_{2}\hbox {O}_{3}\) high refractive index makes it a practical compound in antireflective coatings [6]. When doped with rare-earth ions \((\hbox {Eu}^{3+}, \hbox {Tb}^{3+})\), it presents good luminescence properties [7]. Because of high dielectrics constant and the closest lattice matches to silicon, cubic \(\hbox {Gd}_{2}\hbox {O}_{3}\) has featured as one of the first insulating materials applied for manufacturing of GaAs MOSFETs [1].
At the macroscopic scale cubic \(\hbox {Gd}_{2}\hbox {O}_{3}\) is paramagnetic [8,9,10]. However, there is a difference of opinion about the magnetic properties of low-size \(\hbox {Gd}_{2}\hbox {O}_{3}\). Some of the researchers believe in paramagnetic ordering [10,11,12], while Pederson et al. predicted super paramagnetic behaviors for \(\hbox {Gd}_{2}\hbox {O}_{3}\) nanoparticles [13]. Despite the viewpoints of the former researchers, Ning et al. [14] have emphasized the short-range antiferromagnetic order between the nearest-neighbor Gd ions in the \(\hbox {Gd}_{12}\hbox {O}_{18}\) cluster which is cut from the bulk \(\hbox {Gd}_{2}\hbox {O}_{3}\) crystal. This is a result which is in agreement with the finding of Moon and Kochlcr [15] about first-neighbor Gd ions. But the cubic bulk \(\hbox {Gd}_{2}\hbox {O}_{3}\) in which the long-range magnetic order does occur has been regarded as nearly an ideal paramagnet [15]. Based on our calculation for second- and higher-neighbor Gd ions of cubic bulk \(\hbox {Gd}_{2}\hbox {O}_{3}\), there is no considerable energy difference between ferromagnetic and antiferromagnetic order in the ground state.
The behavior of the bonding electrons in s and p orbitals can be described properly by conventional density functional theory (DFT) using the local density approximation (LDA) [16] and generalized gradient approximation (GGA) [17] approaches. However, those approximations may not describe the highly correlated transition metal d and f electrons correctly. Consequently, it is essential to correct the status of highly correlated d and f electrons separately from the bonding electrons.
For any theoretical illustration of the rare-earth oxides, we must consider the strong on-site f–f interactions which force 4f electrons toward localization.
An efficient, common, easy, and computationally affordable advancement of DFT is the DFT + U method. In this approximation, by adding an empirical on-site Coulomb repulsion potential (U) and Hund exchange coupling constant (J), the lack of correlation in DFT for the 4f orbitals is redefined.
Though the LDA + U [18] or GGA + U [19] are simple and fast methods to include correlation effect compared to other sophisticated ones, they require appropriate Coulomb interaction parameter, U, and Hund exchange coupling constant, J, or equivalently \(U_{\mathrm{eff}}=U-J\). The value of \(U_{\mathrm{eff}}\) which is a physical quantity can be determined through experimental or calculational methods for each compound.
In the present work we compute the electronic and optical properties of cubic \(\hbox {Gd}_{2}\hbox {O}_{3}\) according to the full-potential linearized augmented plane wave (FP-LAPW) method [20].
2 Method of calculation
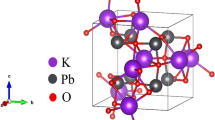
The unit cell of \(\hbox {Gd}_{2}\hbox {O}_{3}\) in the cubic phase is shown in Fig. 1, in which there are two nonequivalent Gd atoms at 8b and 24d sites and one O atom at 48e site [21]. Available experimental lattice constants, together with calculated equilibrium data, are given in Table 1. It is clearly seen that LDA and GGA overestimate the lattice parameter; these findings are consistent with the general tendency of these approximations.

Cubic structure of \(\hbox {Gd}_{2}\hbox {O}_{3}\). Large and small spheres stand for Gd and O atoms, respectively[22]
Calculations were performed to solve Kohn–Sham equations using FP-LAPW method with (LDA) and (GGA) by WIEN2k codes [23]. In order to estimate the U and J values in the DFT + U formalism, there are two major procedures. We can take into account either adjustable phenomenological details or first-principle calculations. The first one is a more practical routine [24, 25], so we preferred to use the value of the Hubbard U very close to that used for the Gd 4f energy levels which are obtained by the positioning of 4f states in the X-ray photo-emission spectra. However, in this procedure we have observed that there is a discrepancy between our result and the band gap found experimentally. Therefore, to use LDA + U and GGA + U, the \(U_{\mathrm{eff}}\) value is chosen 8 eV, as suggested in Ref. [26].

Total energy versus unit cell volume of \(\hbox {Gd}_{2}\hbox {O}_{3}\) in a GGA and b LDA approximation
In the present work, we have considered the configurations of \(\hbox {Gd}_{2}\hbox {O}_{3}\) as Gd:[Xe] \(4f^{7}5d^{1}6s^{2}\) where [Xe] indicates core states and \((4f^{7}5d^{1}6s^{2})\) valence states of Gd atoms, and O: [He] \(2s^{2}2p^{4}\) where [He] indicates core states and \((2s^{2}2p^{4})\) valence states of O atoms. The muffin-tin (MT) spheres radii were chosen to be 2.37 and 1.9 a.u. for Gd and O, respectively. In our calculations, the convergence parameter, \(R_{\mathrm{MT}}K_{\mathrm{max}}\), was set to 9.0 where \(K_{\mathrm{max}}\) gives the magnitude of the largest K vector in plane wave expansion, and \(R_{\mathrm{MT}}\) refers to the smallest of all atomic sphere radii. The lattice harmonics inside the muffin-tin spheres were confined to \(L_{\mathrm{max}} = 10\). Self-consistency is obtained using 3000 k-points in the whole Brillouin zone (BZ), which corresponds to 147 special k-points in the irreducible wedge of Brillouin zone (IBZ). Because of the screened conduction electrons potential of \(\hbox {Gd}_{2}\hbox {O}_{3}\), spin-orbit effects have a negligible effect, so we did not consider spin-orbit interaction in our calculation. The iteration process was executed until the total energy converged to 0.0001 electron charge.
3 Results and discussion
3.1 Optimization of structure
The calculation was performed by assuming at first the experimental lattice constants. Next, the ratio of the total energy of crystal to its primitive unit cell volume (volume optimization) minimized using Murnaghan’s equation of state [27], as shown in Fig. 2. Afterward equilibrium structural parameters were obtained. In Table 1 the calculated equilibrium lattice constants, bulk modulus \(B_{0,}\) and pressure derivative \(B'_{0,}\) are displayed together with available experimental data. It is seen that the GGA values are in agreement with the experimental information.
3.2 Electronic properties
Solid electronic characteristics can be predicted by electrons energy distribution of valance and conduction bands. To describe accurately the contribution of the orbitals that result in the electronic structure of orbitals in electronic structures accurately, the calculated total and partial DOS of \(\hbox {Gd}_{2}\hbox {O}_{3}\) within the LDA + U and GGA + U approaches are presented in Figs. 3, 4, and 5.
As it is seen, the O-2s and Gd–5p states are mainly situated at around −20 to −15 eV. In the energy ranges of about −3 eV to 0 eV, O-2p and Gd-5d states are formed bands on the whole with a width of about 3.25 and 3.3 eV for LDA + U and GGA + U approaches, respectively. Moreover, Gd-5d state stands up at the bottom of the conduction band.

Calculated total and partial DOS of \(\hbox {Gd}_{2}\hbox {O}_{3}\). Fermi energy is aligned to zero

Calculated total DOS of \(\hbox {Gd}_{2}\hbox {O}_{3}\). Fermi energy is aligned to zero

Band structure of \(\hbox {Gd}_{2}\hbox {O}_{3}\) using the LDA approximation. a Down and b up spins

Calculated a real and b imaginary parts of dielectric function for \(\hbox {Gd}_{2}\hbox {O}_{3}\)
Whereas the maximum of the highest occupied orbitals and the minimum of the lowest unoccupied orbitals both occur at the \({\Gamma }\)-point, as illustrated in Fig. 6, \(\hbox {Gd}_{2}\hbox {O}_{3}\) is a direct-gap insulator. Indeed, the forbidden energy gap of \(\hbox {Gd}_{2}\hbox {O}_{3}\) depends on the energy of Gd-5d and O-2p antibonding level. The calculated band gap was found to be 3.61 eV for LDA + U and 3.59 eV for GGA + U method. These values are smaller than the experimental band gap 4.9–5.8 eV, but are in agreement with theoretically calculated value published by Zhang et al. [28]. The observed discrepancy between the experimental and calculational band gap could be caused by polaronic effect. It is well known that in many of the rare-earth compounds due to their ionic nature and localized charge, polaron is formed. So the self-energy of the electronic polaron must be taken into account in pseudopotential calculations, while in the standard LDA or GGA the polaron self-energy is not considered [29, 30].
Table 2 gives a summary of the cubic Gd2O3 band gap values, either calculated in this work or obtained by other methods.
3.3 Optical properties
Measurements of solid optical properties are important in both basic research and industrial applications. To understand the optical properties of materials, we need to study the dielectric function which describes the response of matter to incident electromagnetic fields. The dielectric function \(\epsilon \,({\omega })\) is given by
The imaginary part of the dielectric function \(\epsilon _{2}\,(\omega )\) is given by [34]
The dielectric function imaginary part is connected to the joint DOS and the momentum matrix elements.
To compute the direct interband contribution to the imaginary part of dielectric function \(\epsilon \,({\omega })\), the sum of all possible electron transitions from occupied to unoccupied states must be considered. To obtain the real part of dielectric function \(\epsilon _{1}\,(\omega )\), one can use Kramers–Kronig relation.
The electron energy-loss function [35]
is another valuable tool for probing various properties of materials, which describes the energy loss of fast electrons traversing the material. Sharp maxima of this function are due to plasma oscillations.
An approximation for the oscillator strengths distribution for both intraband and interband transitions could be obtained by the sum rule. The sum rule can be applied to determine some quantitative parameters same as the effective number of valence electrons. The effective number of valence electrons per unit cell that contribute to transitions with frequencies lower than \(\omega \) is given by [34]:
The calculated imaginary and real parts of the dielectric function for \(\hbox {Gd}_{2}\hbox {O}_{3}\) in LDA + U and GGA + U methods are shown in Fig. 6 for energy magnitude up to 50 eV. The variation range of dielectric function indicates that \(\hbox {Gd}_{2}\hbox {O}_{3}\) might be appropriate for high-frequency UV device applications.
Three spectral regions can be separated in Fig. 6. The first region which stretches to about 25 eV specified by a sharp structure connected with valence to conduction band transitions. The second region which is specified by a rapid decrease in the reflectance stretches between 25 and 35 eV. Because the reflectance rises in the third region, the optical absorption occurs again. This action is related to transitions between filled d bands and the empty conduction band states.
Peaks that appeared in the imaginary part of dielectric function are due to energy transition between some orbital. In the curve \(\epsilon _{2}\,({\omega })\), it is obvious that there is an intense absorption peak in the energy range of 3.4–17.4 eV. The first peaks appear from 4 to 8.8 eV. In comparison with Fig. 3, we find they originate from O-2p valence bands to Gd-5d conduction band transitions on the whole. Next to the main peaks, the lowest peaks appear at 19.4 eV, which result in from direct transitions between O-2s and Gd-5d orbitals. Also the next peaks at 23 eV are related to direct transitions between O-2s and Gd-5d orbitals. At the higher photon energy, spectrum decays very rapidly and there is no subject matter.

\(\hbox {Gd}_{2}\hbox {O}_{3}\) refractive index (n) and extinction coefficient (k)
Other important optical functions, including the refractive index n \((\omega )\) and the extinction coefficient k \((\omega )\), could be computed by the real and the imaginary parts of the dielectric function. The extinction coefficient and refractive index are displayed in Fig. 7. The refractive index is almost constant for lower energies, and as the energy increases, it gains a maximum value and exhibits a diminution trend for higher energy values. The static refractive index n (0) is found to have the values of 2.08 and 2.03 for LDA + U and GGA + U methods, respectively, which are in reasonable accord with the value of 1.976 obtained by other methods [35]. It rises with energy in the transparent region and reaches up a highest point in the ultraviolet area at 4.9 eV. The refractive index is closely related to bonding; however, any mechanism which rises electron density in a material also grows up refractive index. The static refractive index value for C-type \(\hbox {Gd}_{2}\hbox {O}_{3}\) is calculated in this study, and values obtained from other techniques are brought together in Table 3.
The optical conductivity of \(\hbox {Gd}_{2}\hbox {O}_{3}\) in terms of energy was computed and is shown in Fig. 8. Evidently, an exciton is a bound state of an electron and an electron hole, moving in the periodic structure of a crystal; hence, its displacement through the crystal gives rise to the transportation of energy but not charge [37]. An exciton can be formed when a photon is absorbed by a crystal and excites an electron from valence band into conduction band. Insulators contain bound electron–hole pairs called an excitonic insulator. The optical conductivity curve in Fig. 8 shows us such an excitonic appearance at 9.4–10.35 and 26.29–27.4 eV. These peaks are due to electron transitions from O-2p and O-2s valence bands to Gd-5d conduction bands.

Optical conductivity spectrum of \(\hbox {Gd}_{2}\hbox {O}_{3}\)
In Fig. 9, the electron energy-loss spectrum is plotted for \(\hbox {Gd}_{2}\hbox {O}_{3}\) in the cubic phase. The peaks of the electron energy-loss spectrum have different origins, such as interband excitations, intraband excitations, or charge carrier plasmons. The first peak located at 14.95 eV originates from O-2p to Gd-5d orbitals. The real parts of the dielectric function at point \((\epsilon _1 (\omega )=0)\) may indicate a plasma resonance. By considering the \(\epsilon _{1}\,(\omega )\) spectrum, we find that \(\epsilon _{1}\,(\omega )=0\) at 31.5 eV, which corresponds to the plasmon peak in L \((\omega )\). The maximum peak of L \((\omega )\) energy at 32.15 eV is allocated to the volume plasmon energy, \(h{\omega }_{p}\). It may attribute to electron excitation from O-2s orbital to the lower and upper conduction bands.

Electron energy-loss spectrum of \(\hbox {Gd}_{2}\hbox {O}_{3}\)
The effective number of valance electrons for \(\hbox {Gd}_{2}\hbox {O}_{3}\) is shown in Fig. 10. Up to 5 eV (below the band gap) there is no electron; then, it rises rapidly and saturates with a value of 210 for the effective number of electrons at about 40 eV. However, in this transition, the main contribution to \(N_{\mathrm{eff}}\) is caused by O-2p bands, in the next levels followed by Gd-5d, Gd-6p, and O-2s band, which is the least.

The effective number of valance electrons for \(\hbox {Gd}_{2}\hbox {O}_{3}\)
4 Conclusion
In this work, we have studied the electronic and optical properties of \(\hbox {Gd}_{2}\hbox {O}_{3}\) in cubic structure symmetry within the framework of density functional theory using the LDA + U and GGA + U methods. Calculations show that the added effective potential, \(U_{\mathrm{eff}}\), provides an enhanced description of the orbital’s location. A direct band gap at the \({\Gamma }\) direction is found for both LDA + U and GGA + U approaches. Calculated GGA values are in good agreement with obtainable experimental data. Analysis of the dielectric function revealed that \(\hbox {Gd}_{2}\hbox {O}_{3}\) compound might be appropriate for high-frequency UV device applications.
References
Hong, M., Kwo, J., Kortan, A.R., Mannaerts, J.P., Sergent, A.M.: Epitaxial cubic gadolinium oxide as a dielectric for gallium arsenide passivation. Science 283, 1897 (1999)
Menzler, N.H., Tietz, F., Uhlenbruck, S., Buchkremer, H.P., Stover, D.: Materials and manufacturing technologies for solid oxide fuel cells. J. Mater. Sci. 45, 3109 (2010)
Sammes, N., Du, Y.: Intermediate-temperature SOFC electrolytes. Fuel Cell Technol. State Perspect. NATO Sci. Ser. 202, 19–34 (2005)
Eyring, L.: The binary rare earth oxides. Handb. Phys. Chem. Rare Earths 3, 337 (1979)
Bonnet, G., Lachkar, M., Larpin, J.P., Colson, J.C.: Characterization of thin solid films of rare earth oxides formed by the metallo-organic chemical vapour deposition technique, for high temperature corrosion applications. Thin Solid Films 31, 261 (1995)
Heitmann, W.: Reactively evaporated films of scandia and yttria. Appl. opt. 12, 394 (1973)
Zych, E.: On the reasons for low luminescence efficiency in combustion-made \(\text{ Lu }_{2}\text{ O }_{3}\): Tb. Opt. Mater. 16, 445 (2001)
Paul, S., Gangopadhyay, A., Sarkar, A.: Characterization of \(\text{ Gd }_{2}\text{ O }_{3}\) nanoclusters. Condens. Matter Mater. Phys. 665, 127–131 (2013). (Book Series: Advanced Materials Research)
Jacobsohn, L.G., Bennett, B.L., Muenchausen, R.E., Tornga, S.C., Thompson, J.D., Ugurlu, O., Cooke, D.W., Lima Sharma, A.L.: Multifunction \(\text{ Gd }_{2}\text{ O }_{3}:\text{ EuGd }_{2}\text{ O }_{3}\): eu nanocrystals produced by solution combustion synthesis: structural, luminescent, and magnetic characterization. J. Appl. Phys. 103, 104303 (2008)
Jamnezhad, H., Jafari, M.: Structure of \(\text{ Gd }_{2}\text{ O }_{3}\) nanoparticles at high temperature. J. Magn. Magn. Mater. 408, 164–167 (2016)
Fortin, M.A., Petotal Jr., R.M., Söderlind, F., Klasson, A., Engström, M., Veres, T., Käll, P.O., Uvdal, K.: Polyethylene glycol-covered ultra-small \(\text{ Gd }_{2}\text{ O }_{3}\) nanoparticles for positive contrast at 1.5 T magnetic resonance clinical scanning. Nanotechnology 18, 395501 (2007)
Mutelet, B., Keller, N., Roux, S., Flores-Gonzales, M.A., Lux, F., Martini, M., Tillement, O., Billotey, C., Janier, M., Villiers, C., Novitchi, G., Luneau, D., Perriat, P.: Size-induced effect upon the Néel temperature of the antiferro/paramagnetic transition in gadolinium oxide nanoparticles. Appl. Phys. A 105, 215–219 (2011)
Pedersen, H., Ojamae, L.: Towards biocompatibility of \(\text{ RE }_{2}\text{ O }_{3}\) nanocrystals—water and organic molecules chemisorbed on \(\text{ Gd }_{2}\text{ O }_{3}\) and \(\text{ Y }_{2}\text{ O }_{3}\) nanocrystals studied by quantum-chemical computations. Nano Lett. 6, 2004–2008 (2006)
Ning, L., et al.: Density functional theory study of magnetic coupling in the \(\text{ Gd }_{12}\text{ O }_{18}\) cluster. J. Phys. Chem. A 112(51), 13650–13654 (2008)
Moon, R.M., Koehler, W.C.: Magnetic properties of \(\text{ Gd }_{2}\text{ O }_{3}\). Phys. Rev. B 11(4), 1609 (1975)
Wu, Z., Cohen, R.E.: More accurate generalized gradient approximation for solids. Phys. Rev. B 73, 235116 (2006)
Perdew, J.P., Burke, K., Ernzerhof, M.: generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996)
Anisimov, V.I., Zaanen, J., Andersen, O.K.: Band theory and Mott insulators: Hubbard U instead of Stoner. Phys. Rev. B 44, 943 (1991)
Anisimov, V.I., Solovyev, I.V., Korotin, M.A., Czyżyk, M.T., Sawatzky, G.A.: Density-functional theory and NiO photoemission spectra. Phys. Rev. B 48, 16929 (1993)
Shick, A.B., Liechtenstein, A.I., Pickett, W.E.: Implementation of the LDA+U method using the full-potential linearized augmented plane-wave basis. Phys. Rev. B 60, 10763 (1999)
Kennedy, B.J., Avdeev, M.: The structure of C-type \(\text{ Gd }_{2}\text{ O }_{3}\). A powder neutron diffraction study using enriched \(^{160}\)Gd. Aust. J. Chem. 64, 119–121 (2011)
Zinkevich, M.: Thermodynamics of rare earth sesquioxides. Prog. Mater. Sci. 52, 597–647 (2007)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2k, an Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. Vienna University of Technology, Vienna (2001)
Anisimov, V.I., Aryasetiawan, F., Lichtenstein, A.I.: First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA+ U method. J. Phys. Condens. Matter 9(4), 767 (1997)
Raekers, M., et al.: Electronic and magnetic structure of R ScO 3 (R= Sm, Gd, Dy) from x-ray spectroscopies and first-principles calculations. Phys. Rev. B 79(12), 125114 (2009)
Larson, P., Lambrecht, W.R.L., Chantis, A., van Schilfgaarde, M.: Electronic structure of rare-earth nitrides using the LSDA+U approach: importance of allowing 4f orbitals to break the cubic crystal symmetry. Phys. Rev. B 75, 045114 (2007)
Murnaghan, F.D.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 30, 244 (1944)
Zhang, X., Lang, M., Wang, J.W., Becker, U., Ewing, R.C.: Structural phase transitions of cubic \(\text{ Gd }_{2}\text{ O }_{3}\) at high pressures. Phys. Rev. B 78, 064114 (2008)
Lal, H.B., Gaur, K.: Electrical conduction in non-metallic rare-earth solids. J. Mater. Sci. 23(3), 919–923 (1988)
Devreese, J.T.: “Polarons” Encyclopedia of Applied Physics, vol. 14. Wiley-VCH Publishers, Inc, Weinheim (1996)
Sahoo, N.K., Senthilkumar, M., Thakur, S., Bhattacharyya, D.: Correlation of optical and microstructural properties of \(\text{ Gd }_{2}\text{ O }_{3}\) thin films through phase-modulated ellipsometry and multi-mode atomic force microscopy. Appl. Surf. Sci. 200, 219 (2002)
Patra, C.R., Mastai, Y., Gedanken, A.: Microwave-assisted synthesis of submicrometer GaO (OH) and \(\text{ Ga }_{2}\text{ O }_{3}\) rods. J. Nanopart. Res. 6, 509 (2004)
Badylevich, M., Shamuilia, S., Afanas’ev, V.V., Stesmans, A., Laha, A., Osten, H.J., Fissel, A.: Investigation of the electronic structure at interfaces of crystalline and amorphous \(\text{ Gd }_{2}\text{ O }_{3}\) thin layers with silicon substrates of different orientations. Appl. Phys. Lett. 90, 252101 (2007)
Ambrosch-Draxl, C., Sofo, J.O.: Linear optical properties of solids within the full-potential linearized augmented plane wave method. Comput. Phys. Commun. 175, 1–14 (2006)
Hummel, Rolf E.: Electronic Properties of Materials. Springer, Berlin (2011)
Liu, Liqin, Chen, Xueyuan: Energy levels, fluorescence lifetime and Judd–Ofelt parameters of Eu\(^{3+}\) in Gd\(_{2}\)O\(_{3}\) nanocrystals. Nanotechnology 18, 255704 (2007)
Liang, W.Y.: Excitons. Phys. Educ. 5(4), 226 (1970)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jamnezhad, H., Jafari, M. Structural, electronic, and optical properties of C-type \(\hbox {Gd}_{2}\hbox {O}_{3}\): a density functional theory investigation. J Comput Electron 16, 272–279 (2017). https://doi.org/10.1007/s10825-017-0974-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10825-017-0974-y