
Abstract
Identifying the viability of protein targets is one of the preliminary steps of drug discovery. Determining the ability of a protein to bind drugs in order to modulate its function, termed the druggability, requires a non-trivial amount of time and resources. Inability to properly measure druggability has accounted for a significant portion of failures in drug discovery. This problem is only further exacerbated by the large sample space of proteins involved in human diseases. With these barriers, the druggability space within the human proteome remains unexplored and has made it difficult to develop drugs for numerous diseases. Hence, we present a new feature developed in eFindSite that employs supervised machine learning to predict the druggability of a given protein. Benchmarking calculations against the Non-Redundant data set of Druggable and Less Druggable binding sites demonstrate that an AUC for druggability prediction with eFindSite is as high as 0.88. With eFindSite, we elucidated the human druggability space to be 10,191 proteins. Considering the disease space from the Open Targets Platform and excluding already known targets from the predicted data set reveal 2731 potentially novel therapeutic targets. eFindSite is freely available as a stand-alone software at https://github.com/michal-brylinski/efindsite.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pharmacology exploits the ability of bioactive compounds to bind with a sufficient specificity to macromolecular targets modulating their functions. New pharmaceuticals are developed through the onerous and often expensive process of drug discovery. In 2010 the overall cost of developing a drug and bringing it to market was estimated at 1–2 billion dollars with a 14-year cycle [1]. In 2016, research done by the Tufts Center for the Study of Drug Development put the cost of bringing a drug to market at $2.6 billion with nearly 11.3% of drugs that enter clinical testing being ever be approved in the United States [2]. This is down from 16.4% of drugs in 2005 [3]. Therefore, technology must be developed to increase accuracy and precision in order to reduce costs and miss-rates in drug discovery. It should be emphasized that most known proteins binding small molecules have no known confirmed therapeutic effect. Although the ChEMBL database comprises around 5000 known proteins with bindable pockets [4], only around 700 of these proteins are confirmed therapeutic targets for FDA approved drugs [5]. Consequently, the candidacy of a protein, and thus a protein pocket, for drug discovery is incredibly hard to confirm.
A large portion of the cost of drug development is due to developmental failures. It is estimated that nearly 60% of drug discovery failures are due to invalid or inappropriate identification of drug targets [6]. This is in part due to the large discovery space of possible drug-protein interactions. The total chemical space of drug discovery is estimated to be 1060 possible compounds [7], which can be alleviated somewhat with chemical fragment libraries and physiochemical thresholds reducing this number to roughly 1023 possible drugs [8]. Drug discovery is further complicated by the large number of possible protein targets for therapeutics in humans. Estimates put the portion of the human proteome related to some disease pathology at 10% [9, 10]. According to the Open Targets Platform this number appears as a conservative estimate because nearly 25,000 human proteins are within the top 5th percentile of disease association scores [11]. With these challenges, it can be concluded that the validation of drug targets in an experimental setting is logistically intensive.
In drug discovery, the analysis of a protein druggability is integral to successful target validation. Druggability, termed over 15 years ago [10], is currently defined as the ability of a protein to be modulated by small drug-like molecules, defined by Lipinski’s Rule of Five [12], with sufficient affinity and specificity in vivo to create a therapeutic effect in a relevant cellular pathway [1]. Traditionally, druggability was analyzed by co-crystalizing proteins with organic solvents to expose possible hydrophobic pockets [13]. This approach eventually evolved to the use of high-throughput screens and nuclear magnetic resonance (NMR) analysis of chemical fragment libraries [14]. In turn, hit rates were used as a metric for protein druggability. However, these methods were problematic as they had low sensitivity and high protein consumption [15]. Recent approaches such as fragment-based NMR fluorescence assays work to overcome these problems [16]. Despite advancements in NMR spectroscopy, experimental methods are still problematic in that their accuracies are directly linked to the fragment library being used. Negative results from drug targets are generally inconclusive and can only be controlled for using more complex and diverse libraries. The same problem extends to reproducibility as the results of these tests are not normalized across fragmentation libraries [17]. In response, the wide availability of pharmacologically relevant data sets has allowed many groups to turn to computationally driven solutions to assessing druggability.
In silico analysis of druggability starts with building models of drug binding pockets. Pocket prediction of in the past has heavily relied on the high-resolution structural data from X-ray crystallography and NMR spectroscopy. The effort and time needed to produce such data is non-trivial even with new methods emerging such as cryo-Electron Microscopy (cryo-EM). Even among known drug targets, a portion of the proteome heavily overrepresented in structural biology, only half of the structures have been elucidated [18]. To overcome the lack of high-resolution data, researchers have started turning to sequence-based homology modeling to develop accurate protein pocket and ligand prediction software. Homology modeling has a discrete advantage in that nearly 95% of known drug targets are represented by an acceptable homolog thus increasing the overall coverage of pharmacologically relevant protein structures [18]. In this paper, eFindSite [19, 20] is used to develop a new druggability classifier. eFindSite employs meta-threading to detect weakly homologous templates, clustering techniques, supervised and unsupervised machine learning, and a confidence estimation system to accurately predict drug-binding pockets in protein structures. eFindSite thus provides a convenient means of pocket detection that can reliably analyze the protein without the need of high-resolution data.
Many druggability classifiers rely on the use of geometric and physiochemical descriptors to predict protein pocket druggability. Geometric descriptors involve the size and complexity of the cavity with the hypothesis that they are directly correlated with easier drug binding and thus higher druggability. However, these descriptors are heavily dependent on the structural information from the pocket prediction algorithm being used. Thus, there is usually weak correlation between different data sets involving these descriptors. Unlike geometric descriptors, however, the significance of physiochemical descriptors of the pocket have been found to be generally independent of the accuracy of the pocket prediction algorithm [21]. Typically, druggability models look for closed hydrophobic pockets within a protein target. These models lean on the knowledge that electrostatic interactions between the ligand and the drug are in opposition to the desolvation energies. In a low dielectric medium such as one exemplified by a lipophilic pocket, the electrostatic interactions are heightened in a quantifiable way [22]. Thus, hydrophobicity as described by [23] is a prominent feature in assessing druggability. Due to these effects, it has been hypothesized that polar residues matter significantly in context specific instances as they often act as hydrogen bond donors in drug-target interactions [22]. Another physiochemical characteristic of note is aromaticity, especially in the case of tyrosine and tryptophan residues. Aromatic amino acids have been hypothesized to interact with drugs using cation-π bonding and π-stacking. In the case of tyrosine and tryptophan, the NE1 and OH groups act to enrich the environment of the pocket with hydrogen bonds [24].
In this paper, we present a method to detect druggability of a pocket that conforms to previous physiochemical findings. A machine learning classifier is developed using descriptors from pocket prediction parameters calculated in eFindSite and the characteristics of the active residues of the pocket. Thus, the model is fully embedded into eFindSite to create an all-in-one software for pocket prediction and analysis. Finally, an inspection of the human proteome with eFindSite was done to quantify a portion of feasibly druggable proteins. This is done in hopes to illuminating novel classes of druggable targets that have yet been explored by scientists either due to a lack of existing structural data, or due to the large nature of the drug target space in humans.
Methods
Druggability data set
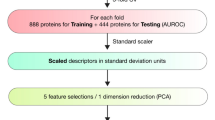
Training machine learning requires training data for the algorithm to analyze and adapt from. The Non-Redundant data set of Druggable and Less Druggable binding sites (NRDLD) provides us with a wholistic analysis on the druggability of over 130 known proteins [25]. This data set, however, lacks proper resolution for our research since it does not specify polypeptide chains of the proteins. To account for heteropolymers, each of the 198 protein chains had their pockets manually analyzed using VMD [26] for druggable ligands. Pockets are labeled as druggable or less druggable based on whether their ligand structures match any known drugs followed by cross referencing against the PDB [27]. The ligands are matched to each pocket using the pocket with the shortest Euclidean distance between the geometric center of the pocket and the geometric center of the ligand. A curated druggability data set with polypeptide chain resolution along with protein pocket ligands confirming pocket druggability is compiled. A threshold of 6 Å Euclidean distance between the pocket center predicted by eFindSite and the geometric center of a ligand is used to make sure that accurately predicted pockets are used in the training set. This modified data set of 240 predicted pockets in 181 polypeptide chains is used to train druggability classifiers.
Feature selection
Two types of descriptors are considered when analyzing possible feature candidates in druggability elucidation. Protein pocket predictors are hypothesized to have an extended application in druggability prediction. Table 1 lists seven pocket descriptors computed by eFindSite chosen as possible feature candidates, including the fraction of templates assigned to a particular pocket (temp_frac), the log of the absolute number of templates assigned to a pocket (temp_log), the average Template Modeling score [28] of the templates to a target (TM-score), the average confidence of binding residues (res_conf), the log of the number of binding residues (res_log), the Protein–Ligand Binding index (PLB_index) [29], and the pocket confidence score (pock_conf).
eFindSite software predicts the relevant residues within the protein pocket and thus physiochemical properties of these binding residues are analyzed as possible druggability feature candidates [19]. Based on findings of previous druggability prediction software, including DrugPred [25] and PockDrug [21], hydropathy, weighted frequency of polar residues (polar_freq), weighted frequency of aromatic residues (aromatic_freq), and frequency of tyrosine atoms (tyr_freq) are included in parameter analysis (Table 1). All frequencies are weighted using confidence estimates calculated by eFindSite.
After the candidates are chosen, violin plots are calculated for the NRDLD druggability data set in order to visualize the distributions of each of the 11 features in druggable and non-druggable proteins. To quantify the correlations and to exclude the possibility of randomness accounting for these correlations, the Monte-Carlo variant of the Fischer-Pittman permutation test is applied via a permute python module [30]. The data set is resampled 100,000 times without replacement to calculate p-values. Any feature with a p-value > 0.02 is discarded from the druggability classifier.
Prediction models
The druggability data set, while reflecting a great deal of research, is relatively small sample size to work with statistically. In choosing the machine learning algorithms, rather than select more popular machine learning models such as neural networks, support vector machines, and random forest techniques, a more basic approach is taken using graphical machine learning models in order to reduce the possibility of overfitting. The two models settled on are logistic regression (LR) and linear discriminant analysis (LDA). A weight vector is calculated via the scikit-learn module [31] for LDA, and the Newton–Raphson method [32] is used for the dichotomous case of LR. Since the Newton Raphson and LDA both require matrix inversions, using all nine pocket descriptors is too unstable for the algorithm. Thus, two sets of parameters are used to create two different classifiers for LDA and LR with different sets of pocket descriptors employed developed to elucidate druggability.
Model evaluation
Classifiers are evaluated using the Receiver Operating Characteristic (ROC) analysis. ROC displays in a plot the fall-out, or false-positive rate (FPR), i.e. the inability to recognize non-druggable pockets, against the sensitivity, or true-positive rate (TPR), i.e. the capacity to correctly identify druggable binding sites. The area under the ROC curve (AUC) is calculated and compared amongst models to establish the default model to be implemented into eFindSite. The confidence intervals of the AUC are estimated non-parametrically by bootstrapping 100,000 times. A 10-fold cross validation is used to confirm the viability of a machine learning model. To optimize threshold values, the Mathew Correlation Coefficient (MCC) [33] is calculated over all possible thresholds between 0 and 1. A model with the highest AUC is used to measure performance against Fpocket, a popular pocket druggability prediction tool with over 250 downloads in the past year alone [34]. This is done using one pocket from each of the 198 polypeptide chains in the NRDLD data set. The model is also independently evaluated for sensitivity on the scPDB data set, a collection of solely druggable proteins across multiple proteomes [35]. The ligands for these proteins have their predicted pockets matched to eFindSite in a manner identical to that of the NRDLD data set. The scPDB data set comprises 15,298 druggable pockets.
Annotated structural human proteome
The structural human proteome is constructed using a reference genome, GRCh38 (Genome Reference Consortium Human Build 38), from the Human genome project [36] downloaded from the Ensembl database [37]. The entire annotated data set comprises 89,872 sequences 50–999 amino acids in length. The 3D structures of these gene products are built with eThread [38] followed by a quality assessment with ModelEvaluator [39] in terms of the estimated Global Distance Test (GDT_TS) score [40]. Subsequently, ligand-binding pockets are predicted in confidently modeled target structures with eFindSite [19]. The top-ranked pockets are subject to fingerprint-based virtual screening [20] against a non-redundant subset of 244,659 small molecules selected from the ZINC library [41]. The druggability of each protein is assessed using the default classifier from eFindSite. The disease space of the human proteome is estimated by mapping gene products to the Open Targets Platform [11], a set of known proteins with significant association to a disease. Those genes with a disease association score of ≥ 0.5 are considered relevant, and any protein expressed from a relevant gene is considered linked, in part, to some disease. Finally, known drug targets in the human proteome are identified by mapping the sequences of gene products to drug targets in DrugBank [42]. Those polypeptides having at least one protein in DrugBank with a sequence identity of ≥ 80% are labelled as known drug targets.
Results and discussion
Binary classifier for pocket druggability
Of the 11 pocket descriptors scrutinized in this study, Table 1 shows that only eight pass the stringent requirement of p-values ≤ 0.02. Five of these descriptors are related to protein pocket prediction, whereas the other three are physiochemical descriptors. The analysis of the distributions of these descriptors is presented in Fig. 1. Due to the unstable nature of computing matrix inversions, the descriptors were organized into two different models. Table 2 shows that the first model (Model 1) introduces a stringent requirement of using descriptors with p-value ≤ 0.001, while the second model (Model 2) uses mainly protein pocket prediction descriptors along with hydropathy. Despite the prevalence of polar attributes in other models [22], the polar_freq value was not statistically relevant to be included in the model. Thus, current models reflect closed “greasy” pockets as the ideal druggable sites.

Violin plots for statistically relevant pocket descriptors. The horizontal blue bar represents the mean, whereas the horizontal purple bar represents the median of a particular data set. The following descriptors are analyzed: atemp_frac, btemp_log, cres_log, dPLB_index, epock_conf, fhydropathy, gtyr_freq, and haromatic_freq
The performances of each of the two LR and LDA models developed in this study are tested on a training data set of 240 protein pockets. Figure 2a shows ROC plots graphed for each of the four classifiers with the AUC of each classifier used to determine its accuracy. The AUC of Model 1 is 0.910 for LR (LR-1) and 0.898 for LDA (LDA-1), whereas the AUC of Model 2 is 0.901 for LR (LR-2) and 0.909 for LDA (LDA-2). The model with the numerically largest AUC (LR-1) is selected as the default classifier for eFindSite. Figure 2b shows that the maximum MCC values of individual models range from 0.6 to 0.7. The default model yields the highest MCC of 0.702 at a probability threshold of 0.732.

Assessment of the performance of draggability classifiers. a ROC plots and b MCC for varying threshold values. Four classifiers are evaluated, LR-1 (Model 1), LR-2 (Model 2), LDA-1 (Model 1), and LDA-2 (Model 2). TPR is the true positive rate and FPR is the false positive rate. Gray regions represent the performance of a random classifier
Fpocket is one of the most widely used protein pocket prediction programs [34]. Due to its geometric approach to modeling pockets and inclusion of relevant polar parameters, Fpocket is the perfect subject of comparison to test the performance of eFindSite. The analysis of all pockets in the NRDLD data set determines AUC values of 0.882 for eFindSite and 0.794 for Fpocket with the corresponding ROC graphs shown in Fig. 3a. Furthermore, 0.95 confidence intervals are 0.845–0.928 for eFindSite and 0.752–0.871 for Fpocket (Fig. 3b). Thus, there is a quantifiable increase in performance from Fpocket to eFindSite.

Performance comparison of eFindSite and Fpocket. This evaluation is conducted against all 198 polypeptides in the curated NRDLD data set. a ROC plots for druggability prediction with eFindSite and Fpocket. TPR is the true positive rate and FPR is the false positive rate. A gray region represents the performance of a random classifier. b Histogram of bootstrapped distributions of randomly resampled AUC scores
Sensitivity validation on the scPDB data set
Sensitivity of the model was independently tested on the scPDB data set. Figure 4 shows that of 15,298 druggable pockets in this set, 9376 pockets are predicted with a high confidence of ≥ 0.8 (solid orange line). Furthermore, the MCC calculated for predicted binding residues is ≥ 0.4 for 12,411 pockets (solid blue line). Encouragingly, as many as 9575 pockets (62.6%) are correctly classified by eFindSite as druggable (solid green line). We may expect the sensitivity accuracy to increase when only confidently and accurately predicted pockets are considered. Indeed, of 9376 confidently predicted pockets, 7667 (81.8%) are classified as druggable (dotted-dashed green line), whereas of 8119 confidently predicted pockets whose MCC is ≥ 0.4, 6727 (82.9%) are classified as druggable (dotted green line). This analysis demonstrates that the sensitivity of druggability prediction with eFindSite is quite high and the majority of confidently identified pockets are in fact druggable.

Performance of eFindSite against the scPDB data set. Histogram of the pocket confidence by eFindSite, the MCC calculated for binding residues predicted by eFindSite, and the druggability calculated with LR-1 (Model 1). Solid lines represent the entire data set, whereas dotted-dashed and dotted lines represent the data set filtered by the pocket confidence and the MCC. Dashed lines mark thresholds at which the data set was filtered
Next, we selected a subset of 101 scPDB proteins whose structures were deposited into the PDB after July 2016 and conducted druggability prediction by eFindSite with a template library constructed from the June 2016 snapshot of the PDB. Figure 5 demonstrates that the performance of eFindSite using a library compiled before the structure of any of target proteins was determined experimentally is only slightly lower than that obtained in February 2017. Indeed, the median druggability values are 0.823 for June 2016 and 0.854 for February 2017 template libraries. Thus, eFindSite is able to function as a prospective predictor.

Violin plots for the retrospective assessment of druggability prediction. The druggability of 101 scPDB proteins, whose structures were deposited into the PDB after July 2016, is predicted by eFindSite with template libraries constructed from June 2016 (purple) and February 2017 (gold) snapshots of the PDB. The horizontal green bar represents the mean, whereas the horizontal red bar represents the median of a particular data set
Druggable pockets in the human proteome
An analysis of druggability is performed on the structural human proteome from the curated GRCh38 data set. Figure 6 shows that 63,713 (70.9%) structure models are confidently predicted with an GDT_TS score of at least 0.4 (solid purple line), of which 39,271 are annotated with putative binding sites by eFindSite. The data set is further pruned for a ≥ 0.8 confidence of the top-ranked pocket resulting in 16,203 (41.3%) proteins used to analyze the druggable human proteome (solid orange line). The druggability of each protein is assessed using the classifier from eFindSite with a probability threshold of ≥ 0.5. From the data set, 10,191 (62.9%) proteins are found to be druggable with high confidence (dotted-dashed green lines).

Inspection of protein druggability across the human proteome. Histogram of the structure confidence assessed with model GDT_TS by eThread, the pocket confidence by eFindSite, and the druggability calculated with LR-1 (Model 1). Solid lines represent the entire data set, whereas dotted-dashed line represents the data set filtered by the pocket confidence. Dashed lines mark thresholds at which the data set was filtered
Analysis of proteins expressed from probable gene disease candidates and known drug targets is conducted to discern relevant proteins for study. Figure 7 shows that the disease space of the human proteome comprises 45,766 gene products (blue circle), whereas 13,412 proteins are known drug targets (red circle). As expected, there is a significant overlap between the disease space and known targets comprising 9357 proteins. Out of 10,191 gene products predicted by eFindSite to be druggable (Fig. 7, green circle), 3867 are already known drug targets, whereas 3593 are outside the disease space according to the Open Targets Platform. Interestingly, as many as 2731 proteins within the predicted druggability space are expressed from genes with disease association scores of ≥ 0.5, yet are not catalogued in DrugBank. These proteins are potentially novel targets that can be exploited for drug discovery.

Analysis of relevant drug targets in the human proteome. The disease space corresponds to those human gene products having a disease association score of ≥ 0.5. Known targets are proteins within the confidently classified data set (GDT_TS by eThread of ≥ 0.4 and the pocket confidence by eFindSite of ≥ 0.8) that have a close homolog in DrugBank (sequence identity of ≥ 0.8). Predicted targets are proteins within the confidently classified data set with a druggability score by eFindSite of ≥ 0.5. The set of all proteins within the disease space and the predicted target space, but not in the known target space are considered relevant novel targets
Case study: α/β hydrolase domain-containing protein 11
Below, we discuss a couple of representative cases selected from the predicted druggable human proteome. Note that neither these proteins nor their close homologs are included in the DrugBank database combining detailed drug data with the comprehensive information on 4985 non-redundant drug targets [42]. The first example is α/β hydrolase domain-containing protein 11 (ABHD11) comprising 315 amino acid residues. A 3D model of ABHD11 was constructed based on the X-ray structure of haloalkane dehalogenase from Xanthobacter autotrophicus (PDB-ID: 2yxp, chain A) [43]. Although both proteins share only 26.3% sequence identity, the estimated GDT_TS score for the ABHD11 model is as high as 0.70. Figure 8a shows the top-ranked pocket (gold) predicted by eFindSite with a 97.4% confidence in the structure model (purple). This binding site comprising 17 residues (H73, G74, L75, F76, F77, H140, S141, M142, F177, Y180, V181, M184, L201, W232, F270, H296, and W297) is assigned a high druggability of 0.98 by the default, LR-1 model.

Druggability assessment of α/β hydrolase domain-containing protein 11 (ABHD11). a Structure model of ABHD11 (solid purple cartoons) with a putative, druggable pocket (a transparent gold surface). Predicted binding residues are shown as solid sticks and the shiny gold sphere is the pocket center. Models of ABHD11 complexed with top-ranked compounds identified with virtual screening: b ZINC63536302 and c ZINC70638822. Small molecules are colored by atom type, relevant pocket residues are represented by solid gold sticks, and target structure is shown in transparent purple
Next, we docked two top-ranked compounds identified by fingerprint-based virtual screening in the ZINC library into the binding site of ABHD11 with eSimDock [44]. The resulting models of ABHD11-ZINC63536302 and ABHD11-ZINC70638822 are shown in Fig. 8b, c, respectively. eSimDock is a similarity-based docking approach that places ligands within the predicted binding sites by superposing them onto ligand-bound templates. It selected the alpha-amino acid ester hydrolase from Acetobacter turbidansand complexed with d-phenylglycine (PDB-ID: 2b4k, chain A, ligand PG9) [45] as a template for the ABHD11-ZINC63536302 model and the human soluble epoxide hydrolase complexed with an inhibitor (PDB-ID: 5all, chain A, ligand II6) [46] for the ABHD11-ZINC70638822 model. Not only are both template proteins structurally similar to ABHD11 with a TM-score of 0.72 (2b4k) and 0.79 (5all), but their bound ligands are also chemically similar to both ZINC compounds with a Tanimoto coefficient (TC) [47] reported by kcombu [48] of 0.39 (PG9 and ZINC63536302) and 0.50 (II6 and ZINC70638822).
An analysis of binding poses of ZINC molecules within the pocket of ABHD11 carried out with the eAromatic program [49] reveals a network of aromatic interactions with the side-chains of Y180, F270, H296, F177, and W297. Moreover, the Ligand Protein Contact (LPC) software [50] reports hydrophobic interactions between the cyclohexoxyl (ZINC63536302) and the 4-hydroxytetrahydropyran (ZINC70638822) moieties, and a cluster of non-polar residues, A202, L206, V209, V215. It is important to note that both compounds selected from the ZINC library by virtual screening closely match the physicochemical parameters of putative binders of ABHD11 estimated by eFindSite, a molecular weight (MW) of 247.0 Da ± 147.5, an octanol–water partition coefficient (logP) of 1.17 ± 2.48, and a polar surface area (PSA) of 78.8 Å2 ± 58.8. The MW, logP, and PSA are, respectively, 291.2 Da, 3.18, and 58.6 Å2 for ZINC63536302, and 263.2 Da, 1.97, and 58.6 Å2 for ZINC70638822.
Case study: 5-aminolevulinic acid synthase 2
Another example of a confidently predicted druggable binding pocket in the human proteome is a putative pyridoxal 5′-phosphate site of erythroid specific mitochondrial 5-aminolevulinate synthase (ALAS2) comprising 587 amino acid residues. Figure 9a shows a 3D model of ALAS2 (purple) constructed based on the X-ray structure of serine palmitoyltransferase from Sphingobacterium multivorum (PDB-ID: 3a2B, chain A) [51]. This model exhibits a modest estimated GDT-score of 0.56 with the 31.6% target-template sequence identity. Figure 9a also shows the top-ranked pocket (gold) predicted by eFindSite with 87.8% confidence comprising 10 residues (C258, F259, H285, A286, S287, H331, S332, V359, H360, and K391). This binding site is assigned a druggability of 0.77 by the LR-1 model.

Druggability assessment of 5-aminolevulinic acid synthase 2 (ALAS2). a Structure model of ALAS2 (solid purple cartoons) with a putative, druggable pocket (a transparent gold surface). Predicted binding residues are shown as solid sticks and the shiny gold sphere is the pocket center. Models of ALAS2 complexed with top-ranked compounds identified with virtual screening: b ZINC00517451 and c ZINC00169159. Small molecules are colored by atom type, relevant pocket residues are represented by solid gold sticks, and target structure is shown in transparent purple
Next, two top-ranked compounds identified by fingerprint-based screening were docked into the binding site of ABHD11 with eSimDock. The constructed models of ALAS2-ZINC00517451 and ALAS2-ZINC00169159 are shown in Fig. 9b, c, respectively. eSimDock selected methionine γ-lyase complexed with β-butenoic acid-pyridoxal-5′-phosphate from Entamoeba histolytica (PDB-ID: 3ael, chain A, ligand 4LM) [52] as the template for both ALAS2-ZINC00517451 and ALAS2-ZINC00169159 models. The template protein has a moderate structure similarity to ALAS2 with a TM-score of 0.46, however, the probability that it shares a pocket with ALAS2 is 0.71. The TC values are 0.66 for 4LM-ZINC00517451 and 0.47 for 4LM-ZINC00169159, indicating sufficiently high chemical similarity to construct reliable template-based complex models.
An analysis with eAromatic shows an aromatic residue, H285, forming parallel stacking with both ligands, whereas LPC reveals hydrophobic interactions between the pyridinyl N1 moiety, and H285 and V359 residues. Further, both compounds selected from the ZINC library by virtual screening have physicochemical parameters similar to the putative binders of ALAS2 estimated by eFindSite: an MW of 254.0 Da ± 123.0, a logP of 0.51 ± 1.14, and a PSA of 122.4 Å2 ± 62.4. The MW, logP, and PSA are, respectively, 167.2 Da, 0.89, and 42 Å2 for ZINC00517451, and 167.2 Da, 1.31, and 42 Å2 for ZINC00169159.
Conclusion
Identification of suitable targets for pharmacotherapy in the human proteome is a critical component of drug development. To improve the state-of-the-art in drug target identification, a new pocket druggability prediction algorithm was developed and implemented in eFindSite. Protein pocket predictors are shown in this study to be generalized to the prediction of pocket druggability. Although certain physiochemical predictors such as the hydropathy and the aromatic character of pocket residues are found to be statistically relevant in the analysis of pocket druggability with current data sets, the pocket polarity is not statistically correlated with druggability. Consequently, the current algorithm favors closed, “greasy” pockets as druggable binding sites.
Subsequently, the extended eFindSite is used to analyze the scope of the druggable human proteome. Our findings indicate that druggable targets make up about 7% of the human proteome. As more data are accumulated, the estimated number of druggable proteins is likely to increase. eFindSite is freely available as a stand-alone software at https://github.com/michal-brylinski/efindsite.
References
Abi Hussein H, Geneix C, Petitjean M, Borrel A, Flatters D, Camproux AC (2017) Drug Discov Today 22(2):404
DiMasi JA, Grabowski HG, Hansen RW (2016) J Health Econ 47:20
Lamberti MJ, Getz KA (2015) White paper: Tufts Center for the Study of Drug Development, Boston, MA
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (2012) Nucleic Acids Res 40(Database issue):D1100
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP (2017) Nat Rev Drug Discov 16(1):19
Brown D, Superti-Furga G (2003) Drug Discov Today 8(23):1067
Bohacek RS, McMartin C, Guida WC (1996) Med Res Rev 16(1):3
Shuker SB, Hajduk PJ, Meadows RP, Fesik SW (1996) Science 274(5292):1531
Edfeldt FN, Folmer RH, Breeze AL (2011) Drug Discov Today 16(7–8):284
Hopkins AL, Groom CR (2002) Nat Rev Drug Discov 1(9):727
Koscielny G, An P, Carvalho-Silva D, Cham JA, Fumis L, Gasparyan R, Hasan S, Karamanis N, Maguire M, Papa E, Pierleoni A, Pignatelli M, Platt T, Rowland F, Wankar P, Bento AP, Burdett T, Fabregat A, Forbes S, Gaulton A, Gonzalez CY, Hermjakob H, Hersey A, Jupe S, Kafkas S, Keays M, Leroy C, Lopez FJ, Magarinos MP, Malone J, McEntyre J, Munoz-Pomer Fuentes A, O’Donovan C, Papatheodorou I, Parkinson H, Palka B, Paschall J, Petryszak R, Pratanwanich N, Sarntivijal S, Saunders G, Sidiropoulos K, Smith T, Sondka Z, Stegle O, Tang YA, Turner E, Vaughan B, Vrousgou O, Watkins X, Martin MJ, Sanseau P, Vamathevan J, Birney E, Barrett J, Dunham I (2017) Nucleic Acids Res 45(D1):D985
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Adv Drug Deliv Rev 46(1–3):3
Ringe D (1995) Curr Opin Struct Biol 5(6):825
Hajduk PJ, Huth JR, Fesik SW (2005) J Med Chem 48(7):2518
Craik DJ, Smith PA, Clark RJ (2010) NMR-based screening and drug discovery. In: Abraham DJ (ed). Burger’s medicinal chemistry and drug discovery. Wiley, Hoboken
Aretz J, Kondoh Y, Honda K, Anumala UR, Nazare M, Watanabe N, Osada H, Rademacher C (2016) Chem Commun (Camb) 52(58):9067
Vukovic S, Huggins DJ (2018) Drug Discov Today 23(6):1258
Somody JC, MacKinnon SS, Windemuth A (2017) Drug Discov Today 22(12):1792
Brylinski M, Feinstein WP (2013) J Comput Aided Mol Des 27(6):551
Feinstein WP, Brylinski M (2014) Mol Inform 33(2):135
Borrel A, Regad L, Xhaard H, Petitjean M, Camproux AC (2015) J Chem Inf Model 55(4):882
Schmidtke P, Barril X (2010) J Med Chem 53(15):5858
Kyte J, Doolittle RF (1982) J Mol Biol 157(1):105
Cammisa M, Correra A, Andreotti G, Cubellis MV (2013) BMC Bioinformatics 14 (Suppl 7):S9
Krasowski A, Muthas D, Sarkar A, Schmitt S, Brenk R (2011) J Chem Inf Model 51(11):2829
Humphrey W, Dalke A, Schulten K (1996) J Mol Graph 14(1):33
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nucleic Acids Res 28(1):235
Zhang Y, Skolnick J (2004) Proteins 57(4):702
Soga S, Shirai H, Kobori M, Hirayama N (2007) J Chem Inf Model 47(2):400
Millman KJ (2015) Permute—a Python package for permutation tests and confidence sets. University of California, Berkeley, 2015
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) J Mach Learn Res 12:2825
Böhning D (1992) Ann Inst Statist Math 44(1):197
Matthews BW (1975) Biochim Biophys Acta 405(2):442
Le Guilloux V, Schmidtke P, Tuffery P (2009) BMC Bioinformatics 10:168
Kellenberger E, Muller P, Schalon C, Bret G, Foata N, Rognan D (2006) J Chem Inf Model 46(2):717
Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, Fulton RS, Kremitzki M, Magrini V, Markovic C, McGrath S, Steinberg KM, Auger K, Chow W, Collins J, Harden G, Hubbard T, Pelan S, Simpson JT, Threadgold G, Torrance J, Wood JM, Clarke L, Koren S, Boitano M, Peluso P, Li H, Chin CS, Phillippy AM, Durbin R, Wilson RK, Flicek P, Eichler EE, Church DM (2017) Genome Res 27(5):849
Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Juettemann T, Keenan S, Laird MR, Lavidas I, Maurel T, McLaren W, Moore B, Murphy DN, Nag R, Newman V, Nuhn M, Ong CK, Parker A, Patricio M, Riat HS, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Wilder SP, Zadissa A, Kostadima M, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Cunningham F, Yates A, Zerbino DR, Flicek P (2017) Nucleic Acids Res 45(D1):D635
Brylinski M, Lingam D (2012) PLoS ONE 7(11):e50200
Wang Z, Tegge AN, Cheng J (2009) Proteins 75(3):638
Zemla A (2003) Nucleic Acids Res 31(13):3370
Sterling T, Irwin JJ (2015) J Chem Inf Model 55(11):2324
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, Maciejewski A, Gale N, Wilson A, Chin L, Cummings R, Le D, Pon A, Knox C, Wilson M (2018) Nucleic Acids Res 46(D1):D1074
Liu X, Hanson BL, Langan P, Viola RE (2007) Acta Crystallogr D Biol Crystallogr 63(Pt 9):1000
Brylinski M (2013) J Chem Inf Model 53(11):3097
Barends TR, Polderman-Tijmes JJ, Jekel PA, Williams C, Wybenga G, Janssen DB, Dijkstra BW (2006) J Biol Chem 281(9):5804
Oster L, Tapani S, Xue Y, Kack H (2015) Drug Discov Today 20(9):1104
Tanimoto TT. An elementary mathematical theory of classification and prediction. IBM Internal Report, 1958
Kawabata T (2011) J Chem Inf Model 51(8):1775
Brylinski M (2018) Chem Biol Drug Des 91(2):380
Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M (1999) Bioinformatics 15(4):327
Ikushiro H, Islam MM, Okamoto A, Hoseki J, Murakawa T, Fujii S, Miyahara I, Hayashi H (2009) J Biochem 146(4):549
Sato D, Shiba T, Karaki T, Yamagata W, Nozaki T, Nakazawa T, Harada S (2017) Sci Rep 7(1):4874
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119524. Portions of this research were conducted with computing resources provided by Louisiana State University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kana, O., Brylinski, M. Elucidating the druggability of the human proteome with eFindSite. J Comput Aided Mol Des 33, 509–519 (2019). https://doi.org/10.1007/s10822-019-00197-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-019-00197-w